
Sakshi Education
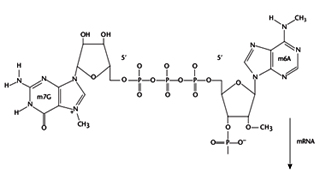
Post Transcriptional Modifications Post transcriptional modifications are absent in prokaryotes. In eukaryotes, the RNA transcripts undergo modifications like splicing, capping and tailing. The messenger ribonucleic acid (mRNA) sequence formed after transcription is exactly similar to one the DNA strand in sequence except that the base uracil is substituted for thymine.
Capping
Addition of cap occurs cotranscriptionally to the 5' end of pre-mRNAs, but is not templated within the DNA. The structure of cap consists of an ‘inverted’ 7-methylated GTP group involving an unusual 5' -ppp-5' linkage with the first templated nucleotide. The main purpose of the cap is to confer stability against 5' exonucleases but is also involved in activating the splicing of 5' proximal introns and activating cap dependent translation initiation.
The cap is added in the following steps:
Capping
Addition of cap occurs cotranscriptionally to the 5' end of pre-mRNAs, but is not templated within the DNA. The structure of cap consists of an ‘inverted’ 7-methylated GTP group involving an unusual 5' -ppp-5' linkage with the first templated nucleotide. The main purpose of the cap is to confer stability against 5' exonucleases but is also involved in activating the splicing of 5' proximal introns and activating cap dependent translation initiation.
The cap is added in the following steps:
- RNA terminal phosphatase (RTPase) removes the terminal phosphate of the 5' triphosphate group of the nascent RNA.
- RNA guanylyl transferase (RGTase) adds GMP to the 5' end using GTP as the donor.
- RNA (guanine-7) methyl transferase adds a methyl group to the N7 position of the cap guanosine, using S-adenosyl methionine as a donor, to produce Cap 0.
- Methyl groups can be added sequentially to the 2' groups of the first and second ribose groups (Cap 1and 2 respectively). If the first templated base is A, it can be methylated on the N6 position, but only if it has first been 2' -O-methylated. The specificity of capping is primarily determined by the RNA polymerase. In addition, only RNAs with a di- or triphosphate at the 5' end are substrates for capping. Therefore products of RNA polymerase I and III, or of endonuclease cleavage, are not capped.
3' end processing
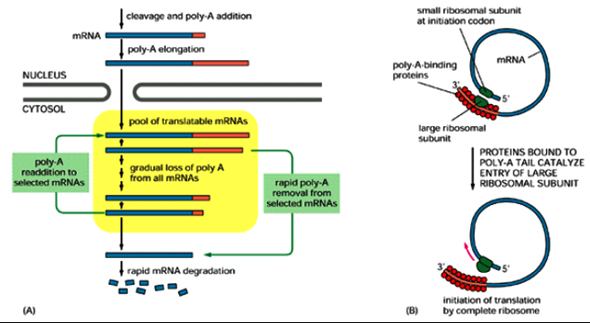
Termination of transcription by RNA polymerase II does not usually occur at fixed positions and often transcribes beyond the 3' end of the mature mRNA and the correct 3' end of mRNAs is generated by a specific endonuclease cleavage followed by non-templated addition of *250 adenosines to form the poly (A) tail. The purpose of addition of poly (A) tail is to confer resistance t0 30' exonucleases and is also essential for efficient translation initiation. The poly (A) tail is added in two steps.
First the RNA is cleaved by an endonuclease, 15–30 nt downstream of a highly conserved hexanucleotide sequence AAUAAA. Poly (A) polymerase then synthesizes the poly A tail using ATP as a substrate. The 3' end processing reaction requires at least two sequences in the RNA. The AAUAAA element provides a binding site for cleavage polyadenylation specificity factor (CPSF), while a region downstream of the cleavage site enriched in U or G and U residues is necessary for binding of cleavage stimulation factor (CstF). Poly(A) polymerase alone synthesizes very long poly A tails, but the presence of poly(A)-binding protein II (PAB II) limits the length of the tail to the physiological length of *250 nt. Histone 3' ends are not polyadenylated but cleaved at their 3 'ends by a meachanism which is unclear.
Splicing:
Introns are intervening sequences that interrupt the sequences that will appear in the mature mRNA (exons) and are usually non coding. However introns like l-19 do code. These are highly reactive and get spliced when RNA assumes the secondary structure after transcription. Therefore these are untranslatable sequences as they remain absent in the mature transcript. Splicing is essentially a process of joining the exons in a pre-mRNA sequence as a consequence of which the introns are eliminated. Introns are present in mostly in in eukaryotes and are rare in prokaryotes. Most of the eukaryotic genes contain introns but genes encoding histones, heat shock proteins do not contain introns. Introns exhibit a wide variation in length and number, with an average size of 3.3 kilobases in humans with an absolute minimum size of 60 bases. Also they vary in number from a few introns up to 177 in the human titin gene. Apart from generating functional mRNAs, splicing assists in the subsequent export of mRNAs to the cytoplasm.
Mechanism of splicing
Intron splicing of pre- mRNA takes place in the nucleus according to a precise and complex arrangement of proteins and ribonuclear particles. Mature mRNA is exported to the cytoplasm for translation. It is a complex process which involves approximately 70 proteins in higher eukaryotes and five small ribonucleoprotein particles (snRNPs). Ultimately pre-mRNAs containing coding sequences (exons) and intervening sequences (introns) are processed to form a continuous mRNA containing only exons. This process is evolutionarily conserved.
Splicing at 5' end introns occurs concomitantly with transcription, whereas 3' introns are largely spliced post-transcriptionally. The RNA elements required for splice-site recognition include sequences at the 5' and 3' splice sites and short consensus sequences such as invariant GU and AG dinucleotides at their 5' and 3' ends respectively and the A at the ‘branchpoint’.
The 5' splice site (sometimes called the ‘donor’ site) has a 9 nucleotide (nt) consensus (A/C)AG/GURAGU, where R denotes a purine and the vertical line represents the exon–intron boundary. The consensus sequence is complementary to a conserved single-stranded region in the abundant U1 (snRNA), and this base-pairing interaction is critical in 5' splice site recognition. The 3' splice site (sometimes called the ‘acceptor’ site) has the consensus CAGG and is preceded by a region enriched in pyrimidines (the polypyrimidine tract). Upstream of the polypyrimidine tract is the ‘branchpoint’ sequence with consensus YNYURAY, where R is a purine, Y is a pyrimidine, N any nucleotide and A is the branchpoint residue. In yeast, there is a much tighter version of this consensus: UACUAAC. The yeast branchpoint sequence is exactly complementary with a conserved single-stranded region of U2 snRNA , allowing for a single mismatch in which the branchpoint A is bulged out. This U2 snRNA-branchpoint interaction is also important in splicing.
There are two types of splicing- Self splicing and Spliceosome mediated splicing
Group I and II introns undergo self splicing.
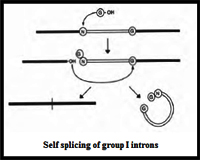
Group I introns are mainly self-splicing introns, also termed as Ribozymes. Splicing of group I introns is processed by two transesterification reactions involving two sequential transfer reactions. The initial reaction is triggered by the attack of an exogenous Guanosine or guanosine nucleotide on the 3'-OH of the phosphodiesdter bond at the 5' splice site resulting in a free 3/-OH group at the upstream exon and the exogenous G being attached to the 5' end of the intron. Then the terminal G of the intron swaps the exoG and occupies the G-binding site to organize the second ester-transfer reaction: the 3'-OH group of the upstream exon attacks the 3' splice site, leading to the ligation of the adjacent upstream and downstream exons and release of the catalytic intron.
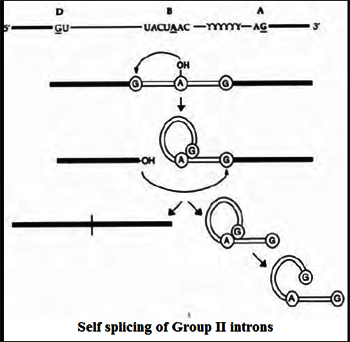
Group II introns are another class of self splicing introns that act as Ribozymes as well as mobile genetic elements. Similar to any splicing mechanism, it also involves two transesterification reactions. Unilke group I introns the first nucleophillic attack is triggered by the 2' OH of the bulged A to attack the 5'-splice site, producing an intron lariat/3'-exon intermediate. In the second step, the 3' OH of the cleaved 5' exon is the nucleophile and attacks the 3'-splice site, resulting in exon ligation and excision of an intron lariat RNA. Some group II introns self-splice in vitro, but the reaction is generally slow and requires nonphysiological conditions—e.g., high concentrations of monovalent salt and/or Mg++ reflecting that proteins are needed to help fold group II intron RNAs into the catalytically active structure for efficient splicing.
Spliceosome mediated splicing:
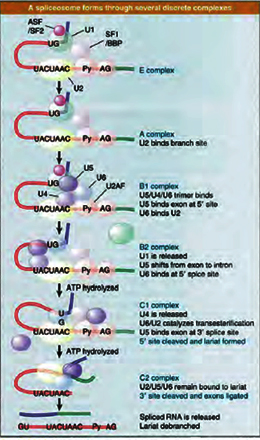
The splicing reaction only occurs after the consensus splice site elements have been recognized by various splicing factors leading to assembly of a ‘Spliceosome’. The spliceosome is a large ribonucleo -protein complex containing 50–100 proteins and five snRNA components (U1, 2, 4, 5 and 6). The snRNAs are each contained within preformed small nuclear ribonucleoprotein (snRNP) complexes (U1, U2, U4/6 and U5), containing several snRNPs, some of which are common to all the spliceosomal snRNPs. U1 and U2 snRNAs are responsible for recognition of the 5' splice site and branchpoint respectively by base pairing. In the fully assembled spliceosome, U2 and U6 snRNAs help to form a network of RNA–RNA interactions that bring together the reactive groups in the pre-mRNA, which are distantly separated in the primary RNA sequence. They also form the catalytic core of the spliceosome, in part by providing specific sites for binding the Mg2+ ions important for catalysis of splicing.
Alternative mRNA Splicing
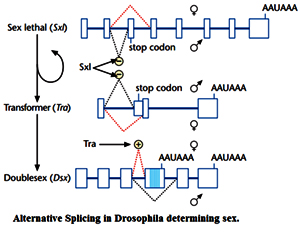
Alternative splicing is the process by which single genes express multiple messenger RNAs (mRNAs), and therefore multiple proteins, by differential processing of the primary transcript (pre-mRNA). The temporal and spatial regulation of protein expression by alternative splicing is determinative for such diverse biological processes as gender specification in Drosophila, commitment to apoptosis, and sound frequency recognition of individual hair cells in the avian cochlea. The first example of alternative splicing was defined in the adenovirus in 1977 and demonstrated that one pre-mRNA molecule could be spliced at different junctions to result in a variety of mature mRNA molecules, each containing different combinations of exons. Shortly afterward, alternative splicing was found to occur in cellular genes as well, with the first example identified in the IgM gene, a member of the immunoglobulin superfamily. Another example of a gene with an impressive number of alternative splicing patterns is the Dscam gene from Drosophila, which is involved in guiding embryonic nerves to their targets during formation of the fly's nervous system. Examination of the Dscam sequence reveals such a large number of introns that differential splicing could, in theory, create a staggering 38,000 different mRNAs. This ability to create so many mRNAs may provide the diversity necessary for forming a complex structure such as the nervous system. In fact, the existence of multiple mRNA transcripts within single genes may account for the complexity of some organisms, such as humans, that have relatively few genes (approximately 20,000).
Trans-splicing
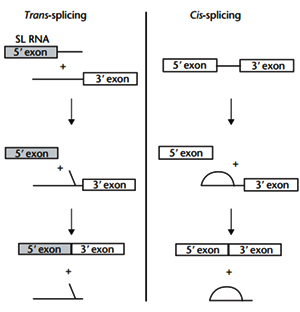
Trans-splicing involves the joining of exons that originate on separate transcripts. Initially, split group II introns in the organelles of plants and algae, and spliced-leader (SL)-dependent trans-splicing in many lower eukaryotes, were the only documented examples of trans-splicing in nature. Since the early 1990s, evidence has been obtained to suggest that trans-splicing may also occur in higher eukaryotes. With the surprisingly low number of identi?ed genes in humans, there has been an interest in determining whether trans-splicing occurs in mammals because this process could potentially increase the coding capacity of a genome. . In this reaction, the 5’ exon is donated from a small RNA polymerase II transcript – the SL RNA. Trans-splicing occurs at a 3’ splice site located on an RNA molecule that is transcribed separately. Addition of SL RNA to a 3’ trans acceptor molecule is not dependent on the formation of base pairs between the two transcripts. The reaction itself is analogous to cis- splicing which involves two sequential trans esterification reactions.
RNA editing:
RNA editing is the only mechanism that can alter the nucleotide sequence of RNA after transcription by addition of nucleotides that are not coded for in the DNA template. RNAediting includes the deamination of cytidines to produce uracil, the deamination of adenosines to produce inosines, or the insertion or deletion of uridine nucleotides. The best characterized types of mRNA editing events in mammalian systems are C to U and A to I editing, which occur by a chemically similar deamination mechanism
A to I editing:
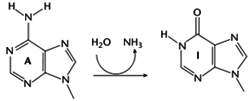
Adenosine can be converted to inosine by hydrolytic deamination at the N6 position). Since I is recognized by the translation machinery as G, this can change the amino acid encoded by the edited codon, although it cannot create new stop codons. For example, in GluRB, an ion channel mRNA, a CAG glutamine codon is edited to CIG, which is recognized as a CGG arginine codon. The template for editing in this case is double-stranded RNA, which forms as a result of complementary sequence in the exon (around the adenosine to be edited) and downstream intron sequence.All RNAs identified to undergo A to I editing are expressed in the central nervous system (e.g. glutamate-gated ion channel receptors (GluR) and serotonin (5-HT2C) receptors). A to I editing is carried out by adenosine deaminases that act on RNA (ADARs).
C to U editing:
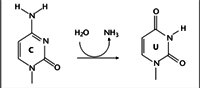
C to U editing occurs in apoB mRNA. ApoB-100, encoded by the unedited mRNA in liver, is a major component of low-density lipoprotein (LDL) and very low-density lipoprotein (VLDL) particles. In the intestine, editing of apoB mRNA at a single cytidine residue (C6666) to U changes a glutamine codon (CAA) to a stop codon (UAA). The resultant truncated protein (apoB-48) lacks the C-terminal LDL receptor binding domain of apoB-100 and is a component of chylomicrons.
C to U editing is catalyzed by APOBEC-1 (apoB mRNA editing enzyme catalytic polypeptide 1), a cytidine deaminase, which in humans is expressed only in the intestine. In contrast to A to I editing, the primary sequence of apoB mRNA is important for editing. The minimal RNA required for specific editing comprises a 5' regulator element (immediately upstream of C6666), a 4 nt spacer element following the edited site, and an 11 nt ‘mooring sequence’ immediately downstream. The mooring site is necessary for high-affinity binding by ACF, which docks APOBEC-1 to the mRNA for specific editing at C6666. ApoB is the only identified.
Termination of transcription by RNA polymerase II does not usually occur at fixed positions and often transcribes beyond the 3' end of the mature mRNA and the correct 3' end of mRNAs is generated by a specific endonuclease cleavage followed by non-templated addition of *250 adenosines to form the poly (A) tail. The purpose of addition of poly (A) tail is to confer resistance t0 30' exonucleases and is also essential for efficient translation initiation. The poly (A) tail is added in two steps.
First the RNA is cleaved by an endonuclease, 15–30 nt downstream of a highly conserved hexanucleotide sequence AAUAAA. Poly (A) polymerase then synthesizes the poly A tail using ATP as a substrate. The 3' end processing reaction requires at least two sequences in the RNA. The AAUAAA element provides a binding site for cleavage polyadenylation specificity factor (CPSF), while a region downstream of the cleavage site enriched in U or G and U residues is necessary for binding of cleavage stimulation factor (CstF). Poly(A) polymerase alone synthesizes very long poly A tails, but the presence of poly(A)-binding protein II (PAB II) limits the length of the tail to the physiological length of *250 nt. Histone 3' ends are not polyadenylated but cleaved at their 3 'ends by a meachanism which is unclear.
Splicing:
Introns are intervening sequences that interrupt the sequences that will appear in the mature mRNA (exons) and are usually non coding. However introns like l-19 do code. These are highly reactive and get spliced when RNA assumes the secondary structure after transcription. Therefore these are untranslatable sequences as they remain absent in the mature transcript. Splicing is essentially a process of joining the exons in a pre-mRNA sequence as a consequence of which the introns are eliminated. Introns are present in mostly in in eukaryotes and are rare in prokaryotes. Most of the eukaryotic genes contain introns but genes encoding histones, heat shock proteins do not contain introns. Introns exhibit a wide variation in length and number, with an average size of 3.3 kilobases in humans with an absolute minimum size of 60 bases. Also they vary in number from a few introns up to 177 in the human titin gene. Apart from generating functional mRNAs, splicing assists in the subsequent export of mRNAs to the cytoplasm.
Mechanism of splicing
Intron splicing of pre- mRNA takes place in the nucleus according to a precise and complex arrangement of proteins and ribonuclear particles. Mature mRNA is exported to the cytoplasm for translation. It is a complex process which involves approximately 70 proteins in higher eukaryotes and five small ribonucleoprotein particles (snRNPs). Ultimately pre-mRNAs containing coding sequences (exons) and intervening sequences (introns) are processed to form a continuous mRNA containing only exons. This process is evolutionarily conserved.
Splicing at 5' end introns occurs concomitantly with transcription, whereas 3' introns are largely spliced post-transcriptionally. The RNA elements required for splice-site recognition include sequences at the 5' and 3' splice sites and short consensus sequences such as invariant GU and AG dinucleotides at their 5' and 3' ends respectively and the A at the ‘branchpoint’.
The 5' splice site (sometimes called the ‘donor’ site) has a 9 nucleotide (nt) consensus (A/C)AG/GURAGU, where R denotes a purine and the vertical line represents the exon–intron boundary. The consensus sequence is complementary to a conserved single-stranded region in the abundant U1 (snRNA), and this base-pairing interaction is critical in 5' splice site recognition. The 3' splice site (sometimes called the ‘acceptor’ site) has the consensus CAGG and is preceded by a region enriched in pyrimidines (the polypyrimidine tract). Upstream of the polypyrimidine tract is the ‘branchpoint’ sequence with consensus YNYURAY, where R is a purine, Y is a pyrimidine, N any nucleotide and A is the branchpoint residue. In yeast, there is a much tighter version of this consensus: UACUAAC. The yeast branchpoint sequence is exactly complementary with a conserved single-stranded region of U2 snRNA , allowing for a single mismatch in which the branchpoint A is bulged out. This U2 snRNA-branchpoint interaction is also important in splicing.
There are two types of splicing- Self splicing and Spliceosome mediated splicing
Group I and II introns undergo self splicing.
Group I introns are mainly self-splicing introns, also termed as Ribozymes. Splicing of group I introns is processed by two transesterification reactions involving two sequential transfer reactions. The initial reaction is triggered by the attack of an exogenous Guanosine or guanosine nucleotide on the 3'-OH of the phosphodiesdter bond at the 5' splice site resulting in a free 3/-OH group at the upstream exon and the exogenous G being attached to the 5' end of the intron. Then the terminal G of the intron swaps the exoG and occupies the G-binding site to organize the second ester-transfer reaction: the 3'-OH group of the upstream exon attacks the 3' splice site, leading to the ligation of the adjacent upstream and downstream exons and release of the catalytic intron.
Group II introns are another class of self splicing introns that act as Ribozymes as well as mobile genetic elements. Similar to any splicing mechanism, it also involves two transesterification reactions. Unilke group I introns the first nucleophillic attack is triggered by the 2' OH of the bulged A to attack the 5'-splice site, producing an intron lariat/3'-exon intermediate. In the second step, the 3' OH of the cleaved 5' exon is the nucleophile and attacks the 3'-splice site, resulting in exon ligation and excision of an intron lariat RNA. Some group II introns self-splice in vitro, but the reaction is generally slow and requires nonphysiological conditions—e.g., high concentrations of monovalent salt and/or Mg++ reflecting that proteins are needed to help fold group II intron RNAs into the catalytically active structure for efficient splicing.
Spliceosome mediated splicing:
The splicing reaction only occurs after the consensus splice site elements have been recognized by various splicing factors leading to assembly of a ‘Spliceosome’. The spliceosome is a large ribonucleo -protein complex containing 50–100 proteins and five snRNA components (U1, 2, 4, 5 and 6). The snRNAs are each contained within preformed small nuclear ribonucleoprotein (snRNP) complexes (U1, U2, U4/6 and U5), containing several snRNPs, some of which are common to all the spliceosomal snRNPs. U1 and U2 snRNAs are responsible for recognition of the 5' splice site and branchpoint respectively by base pairing. In the fully assembled spliceosome, U2 and U6 snRNAs help to form a network of RNA–RNA interactions that bring together the reactive groups in the pre-mRNA, which are distantly separated in the primary RNA sequence. They also form the catalytic core of the spliceosome, in part by providing specific sites for binding the Mg2+ ions important for catalysis of splicing.
Alternative mRNA Splicing
Alternative splicing is the process by which single genes express multiple messenger RNAs (mRNAs), and therefore multiple proteins, by differential processing of the primary transcript (pre-mRNA). The temporal and spatial regulation of protein expression by alternative splicing is determinative for such diverse biological processes as gender specification in Drosophila, commitment to apoptosis, and sound frequency recognition of individual hair cells in the avian cochlea. The first example of alternative splicing was defined in the adenovirus in 1977 and demonstrated that one pre-mRNA molecule could be spliced at different junctions to result in a variety of mature mRNA molecules, each containing different combinations of exons. Shortly afterward, alternative splicing was found to occur in cellular genes as well, with the first example identified in the IgM gene, a member of the immunoglobulin superfamily. Another example of a gene with an impressive number of alternative splicing patterns is the Dscam gene from Drosophila, which is involved in guiding embryonic nerves to their targets during formation of the fly's nervous system. Examination of the Dscam sequence reveals such a large number of introns that differential splicing could, in theory, create a staggering 38,000 different mRNAs. This ability to create so many mRNAs may provide the diversity necessary for forming a complex structure such as the nervous system. In fact, the existence of multiple mRNA transcripts within single genes may account for the complexity of some organisms, such as humans, that have relatively few genes (approximately 20,000).
Trans-splicing
Trans-splicing involves the joining of exons that originate on separate transcripts. Initially, split group II introns in the organelles of plants and algae, and spliced-leader (SL)-dependent trans-splicing in many lower eukaryotes, were the only documented examples of trans-splicing in nature. Since the early 1990s, evidence has been obtained to suggest that trans-splicing may also occur in higher eukaryotes. With the surprisingly low number of identi?ed genes in humans, there has been an interest in determining whether trans-splicing occurs in mammals because this process could potentially increase the coding capacity of a genome. . In this reaction, the 5’ exon is donated from a small RNA polymerase II transcript – the SL RNA. Trans-splicing occurs at a 3’ splice site located on an RNA molecule that is transcribed separately. Addition of SL RNA to a 3’ trans acceptor molecule is not dependent on the formation of base pairs between the two transcripts. The reaction itself is analogous to cis- splicing which involves two sequential trans esterification reactions.
RNA editing:
RNA editing is the only mechanism that can alter the nucleotide sequence of RNA after transcription by addition of nucleotides that are not coded for in the DNA template. RNAediting includes the deamination of cytidines to produce uracil, the deamination of adenosines to produce inosines, or the insertion or deletion of uridine nucleotides. The best characterized types of mRNA editing events in mammalian systems are C to U and A to I editing, which occur by a chemically similar deamination mechanism
A to I editing:
Adenosine can be converted to inosine by hydrolytic deamination at the N6 position). Since I is recognized by the translation machinery as G, this can change the amino acid encoded by the edited codon, although it cannot create new stop codons. For example, in GluRB, an ion channel mRNA, a CAG glutamine codon is edited to CIG, which is recognized as a CGG arginine codon. The template for editing in this case is double-stranded RNA, which forms as a result of complementary sequence in the exon (around the adenosine to be edited) and downstream intron sequence.All RNAs identified to undergo A to I editing are expressed in the central nervous system (e.g. glutamate-gated ion channel receptors (GluR) and serotonin (5-HT2C) receptors). A to I editing is carried out by adenosine deaminases that act on RNA (ADARs).
C to U editing:
C to U editing occurs in apoB mRNA. ApoB-100, encoded by the unedited mRNA in liver, is a major component of low-density lipoprotein (LDL) and very low-density lipoprotein (VLDL) particles. In the intestine, editing of apoB mRNA at a single cytidine residue (C6666) to U changes a glutamine codon (CAA) to a stop codon (UAA). The resultant truncated protein (apoB-48) lacks the C-terminal LDL receptor binding domain of apoB-100 and is a component of chylomicrons.
C to U editing is catalyzed by APOBEC-1 (apoB mRNA editing enzyme catalytic polypeptide 1), a cytidine deaminase, which in humans is expressed only in the intestine. In contrast to A to I editing, the primary sequence of apoB mRNA is important for editing. The minimal RNA required for specific editing comprises a 5' regulator element (immediately upstream of C6666), a 4 nt spacer element following the edited site, and an 11 nt ‘mooring sequence’ immediately downstream. The mooring site is necessary for high-affinity binding by ACF, which docks APOBEC-1 to the mRNA for specific editing at C6666. ApoB is the only identified.
Published date : 02 Jul 2014 05:30PM




More Articles


Most Read
