

Sakshi Education
DNA repair has a fundamental role in DNA metabolism. Its importance in maintaining genome stability not only protects against the onset of malignant transformation but also contributes to the normal development of the organism. In biological systems, two basic types of repair mechanisms exist to repair DNA damage. Some lesions are repaired by cutting out the damaged base or a piece of DNA containing the damaged base and synthesizing new DNA to replace the damaged DNA. This process is called excision repair. Alternatively, other lesions are repaired without the need for synthesizing new DNA. This mechanism involves a process; called Reversal of damage, in which the lesion is physically removed from the base so that the base is restored to its normal appearance.
Direct Repair:
Also known as reversal of damage can be well demonstrated by two well-known examples - UV-induced DNA dimers by photolyases and the repair of mutagenic methyl lesions by methyltransferases
Photolyases:
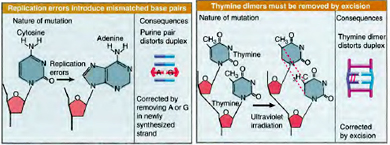
Photolyases use the energy in visible light to split ultraviolet (UV)-induced Cyclobutyl dimmers of cytosine and thymine dimers, in a process called photoreactivation. Photolyases work in concert with two molecular cofactors, or chromophores. The first chromophore captures a photon of visible light and the second chromophore uses the energy of the captured photon to initiate an electron transfer that splits the dimer bonds, restoring the dimerized bases to their normal structure. Photolyases have been found in certain bacteria, yeast and marsupials, but they have not been found in mammals. Photoreactivation is absent in Humans. Cyclobutyl dimers in such organisms lacking photoreactivation are repaired by nucleotide Excision repair.
Methyltransferases:
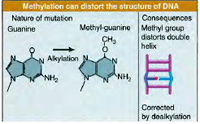 Methyltransferases involved in DNA repair function by removing mutagenic alkyl lesions from oxygen atoms present on the bases guanine (O6-methylguanine) and thymine (O4-methylthymine). Methyltransferases have been found in every type of cell, including bacteria, yeast and humans. Similar to photolyases, these enzymes also work by direct reversal of damage by transferring the methyl group from the DNA to themselves and thereby restoring the base to normal. Subsequent to the transfer, the methyl group remains attached to the protein’s active site, making it inactive. Consequently, each molecule of methyltransferase can repair only one potentially mutagenic lesion. Because of this, methyltransferases are called suicide enzymes to distinguish them from true enzymes, which catalyse multiple reactions.
Methyltransferases involved in DNA repair function by removing mutagenic alkyl lesions from oxygen atoms present on the bases guanine (O6-methylguanine) and thymine (O4-methylthymine). Methyltransferases have been found in every type of cell, including bacteria, yeast and humans. Similar to photolyases, these enzymes also work by direct reversal of damage by transferring the methyl group from the DNA to themselves and thereby restoring the base to normal. Subsequent to the transfer, the methyl group remains attached to the protein’s active site, making it inactive. Consequently, each molecule of methyltransferase can repair only one potentially mutagenic lesion. Because of this, methyltransferases are called suicide enzymes to distinguish them from true enzymes, which catalyse multiple reactions.
Excision repair:
It is of two types- Base excision repair and Nucleotide excision repair.
Base excision repair:
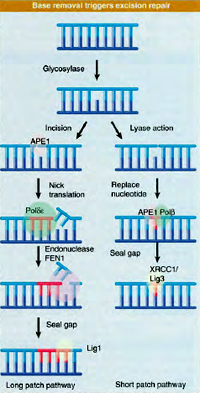 Base excision repair (BER) is a repair mechanism that aims at complete removal of the damaged base from the DNA by excision and is subsequently replaced by a new one.
Base excision repair (BER) is a repair mechanism that aims at complete removal of the damaged base from the DNA by excision and is subsequently replaced by a new one.
In addition, BER is a multistep process that involves more than one protein which can be well explained in E. coli. Initially, an enzyme called DNA glycosylase cleaves the glycosydic bond connecting the sugar and the base resulting in removal of the base. As a consequence, an abasic site or a one-base gap is generated in the DNA helix. Another enzyme called AP endonuclease cuts the sugar–phosphate backbone at the abasic site and the hanging remnants are clipped off by a third enzyme, deoxyribophosphodiesterase. Lastly, a DNA polymerase inserts a new nucleotide in the gap and DNA ligase seals the new nucleotide into the DNA. BER can fix a large number of damaged bases with the aid of different glycosylases which are assigned specific roles to remove a few different damaged bases or types of damaged bases. For instance, uracil DNA glycosylases remove uracils that result from damage to cytosines. The 3-methyladenine DNA glycosylases remove alkylated bases, such as 3-methyladenine, which are not repaired by methyltransferases. Yet another glycosylase removes the oxidatively damaged base 8-oxoguanine. In each case, the glycosylase activity results in an abasic site that is repaired by the rest of the BER pathway. Like the DNA-repair methyltransferases, the BER proteins are found in organisms from bacteria to humans.
Nucleotide excision repair:
 Nucleotide excision repair is of much broader specificity than base excision repair. The major limitation of BER, in spite of its ability to repair a wide range of altered or adducted bases from DNA is that, it can only be used to repair lesions for which there are specific DNA glycosylases. In order to overcome this drawback where the number of lesion types exceed the number of lesion specific glycosylases, cells employ alternate repair mechanisms. There is another process that can repair many different, nonspecific types of base adducts and the repair process is called Nucleotide excision repair (NER). It deals with extreme forms of dmage like intrastrand crosslinks, modofed bases with large chemical groups. NER identifies the DNA damage by recognizing a distortion in the DNA helix caused by the lesion rather than recognizing specific lesions. For example, the pyrimidine dimers caused by ultraviolet light make a bend, or a kink, in DNA molecules. Crosslinks and bulky adducts like the aflatoxin adduct also distort DNA and are substrates for repair by NER. It repairs cyclobutyl dimmers by a dark process without the reuirement of light energy in organisms which like photoreactivation. Unlike in BER, an important observation to be made in NER is that the process does not involve selective base removal but it eliminates an entire piece of DNA containing the lesion, rather than just the damaged base. This complex, multistep repair mechanism can be best explained in E.coli which requires numerous proteins.
Nucleotide excision repair is of much broader specificity than base excision repair. The major limitation of BER, in spite of its ability to repair a wide range of altered or adducted bases from DNA is that, it can only be used to repair lesions for which there are specific DNA glycosylases. In order to overcome this drawback where the number of lesion types exceed the number of lesion specific glycosylases, cells employ alternate repair mechanisms. There is another process that can repair many different, nonspecific types of base adducts and the repair process is called Nucleotide excision repair (NER). It deals with extreme forms of dmage like intrastrand crosslinks, modofed bases with large chemical groups. NER identifies the DNA damage by recognizing a distortion in the DNA helix caused by the lesion rather than recognizing specific lesions. For example, the pyrimidine dimers caused by ultraviolet light make a bend, or a kink, in DNA molecules. Crosslinks and bulky adducts like the aflatoxin adduct also distort DNA and are substrates for repair by NER. It repairs cyclobutyl dimmers by a dark process without the reuirement of light energy in organisms which like photoreactivation. Unlike in BER, an important observation to be made in NER is that the process does not involve selective base removal but it eliminates an entire piece of DNA containing the lesion, rather than just the damaged base. This complex, multistep repair mechanism can be best explained in E.coli which requires numerous proteins.
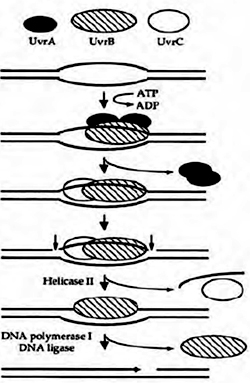
The first step is the detection of DNA damage by a complex of two proteins, UvrA and UvrB. Once the damaged site is identified, Uvr A helps Uvr B bind the damaged region tightly, and then UvrA falls off the DNA. A third protein, Uvr C, joins UvrB on the DNA. Uvr B then
cuts the DNA strand at the fifth phosphodiester bond downstream of the damaged site, on one side of the lesion and UvrC cuts the strand on the other side at the eight phosphodiester bond upstream. A fourth protein, called UvrD, then displaces the twelve nucleotide single-stranded piece of DNA containing the lesion, leaving a single-stranded gap in the double-stranded helix. A DNA polymerase then synthesizes a new tract of single-stranded DNA to fill in the gap,and DNA ligase seals the newly made patch into the DNA. This process of repair mentioned above is known as short patch repair process and is absent in eukaryotes but long patch repair process involving 24-29 nucleotides exists.
In higher organisms, like humans, the process of NER appears to be more complicated, involving even more proteins. But the basic pattern of recognition of damage and removal of a segment of DNA containing the damage is the same. Deficiencies in NER proteins in humans cause several severe inherited diseases.
Mismatch repair
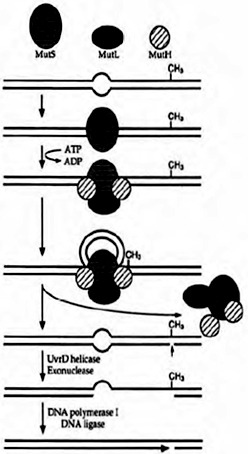 Due to the high processivity of DNA polymerases during replication occasional incorporation of an incorrect or mismatched base opposite a normal base is a common phenonmenon. Mismatches as such do not cause DNA damage, in the sense of an altered or damaged base, but they can nevertheless cause mutations. Therefore cells employ mechanisms to inspect the newly formed DNA after replication for mismatches.
Due to the high processivity of DNA polymerases during replication occasional incorporation of an incorrect or mismatched base opposite a normal base is a common phenonmenon. Mismatches as such do not cause DNA damage, in the sense of an altered or damaged base, but they can nevertheless cause mutations. Therefore cells employ mechanisms to inspect the newly formed DNA after replication for mismatches.
In E. coli, mismatches in the newly replicated DNA are identified and bound by a dimer of Mut S proteins. With the help of a second protein (Mut L), the Mut S dimer binds and activates a third protein, Mut H. Once it is activated, Mut H can make a nick in the newly synthesized DNA strand containing the mismatched base. Other enzymes then move in and remove a segment of the strand containing the incorrect base. This excision step leaves long gaps of up to several thousand bases, which are repaired by a DNA polymerase and DNA ligase. Like NER and BER, mismatch repair is an excision repair process.
An important feature of mismatch repair is that it is targeted to the newly synthesized DNA strand and not on the original (or template) strand which can cause removal of the correct (template)base, instead of the incorrect (new)one. Cells figure out the new strand based on a chemical modification, called Dam methylation, that is present exclusively on the old strand. The MutH enzyme only cuts newly synthesized DNA strands that are missing this modification.
SOS Response:
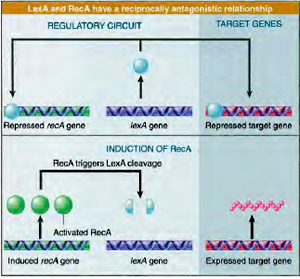 SOS repair when genome is extensively damaged. Cells prefer to replicate thedamaged region in a error prone manner rather than dying. It is a coordinated cellular response which aids in the survival of the organisms by effecting the expression of protein that are involved in cell division, replication, recombination and excision repair. About 30 genes are reported to be involved in this process under normal conditions a protein called Lex A repressor binds to SOS box to repress expression of SOS genes.
SOS repair when genome is extensively damaged. Cells prefer to replicate thedamaged region in a error prone manner rather than dying. It is a coordinated cellular response which aids in the survival of the organisms by effecting the expression of protein that are involved in cell division, replication, recombination and excision repair. About 30 genes are reported to be involved in this process under normal conditions a protein called Lex A repressor binds to SOS box to repress expression of SOS genes.
Under conditions of extensive DNA damage or blockage of replication, formation of large single stranded DNA regions occurs, which are recognised by Rec A protein. Single stranded DNA- Rec A complex bind to Lex A and inactivate the repressor which results in the transcription of SOS genes. As a result of this, cell division stops, excision and recombination activities increase. Replication occurs extensively to bypass the damage. SOS response enables the cell either to eliminate the DNA damage or to survive with damaged DNA.
Recombinational Repair:
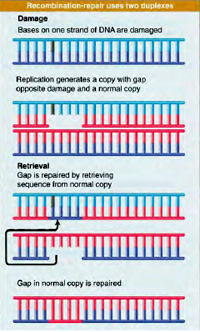 Recombinational repair is a post replication repair process. The recombinational repair pathway responds to the potentially lethal formation double standed DNA breaks (DSBs) that occur in the genome through the direct action of ionizing radiation, exposure to certain chemicals, or the product of blocked replication forks. There are actually three DSB repair pathways that operate in the cell, and defects in each of these pathways have serious consequences for human health. The mechanism of two of the pathways uses homologous recombination, whereas the other does not.
Recombinational repair is a post replication repair process. The recombinational repair pathway responds to the potentially lethal formation double standed DNA breaks (DSBs) that occur in the genome through the direct action of ionizing radiation, exposure to certain chemicals, or the product of blocked replication forks. There are actually three DSB repair pathways that operate in the cell, and defects in each of these pathways have serious consequences for human health. The mechanism of two of the pathways uses homologous recombination, whereas the other does not.
Nonhomologous end rejoining
Nonhomologous end rejoining (NHEJ) is effected without the need for extensive homology between the DNA ends that are to be joined, and it has been considered the principal pathway for DSB repair in mammalian cells. This pathway also processes the site-speci?c DSBs created during variant (diversity) joining (V(D)J) recombination – an essential component for the developmental maturation of the immune system. The mechanism of end rejoining involves the Ku70/80 heterodimer that binds the ends of the double-stranded DNA to be rejoined. This activates the catalytic subunit of DNA-PK, a protein kinase that stabilizes the interaction with the DNA ends. Rejoining is then carried out by the DNA ligase IV/XRCC4 heterodimer.
Homologous recombination and single-strand annealing:
Two pathways use homologous recombination (HR) as a mechanism for DSB repair. HR uses the RAD52 group of proteins to form and resolve Holliday junctions. Mammalian cells carry out HR during mitotic recombination and repair DSBs by HR in late S and G2 phases of the cell cycle, where an undamaged sister chromatid is available. The human breast cancer susceptibility genes breast cancer 1, early onset (BRCA1) and breast cancer 2, early onset (BRCA2) are involved in recombinational repair together with RAD51 homolog (RAD51). In cells with damaged DNA, these three proteins relocate to regions that contain PCNA at sites of DNA damage. ATM, which is mutated in the radiation sensitive disorder ataxia telangiectasia, is proposed to be a damage sensor of DSBs in the HR pathway. ATM phosphorylates BRCA1, which triggers the response to ionizing radiation in mammalian cells.The single-strand annealing (SSA) pathway is a subpathway of HR, which relies on regions of homology to align the strands of DNA to be rejoined. The proteins involved in the pathway are RAD50, MRE11 and NBS1. MRE11, a 30–50 exonuclease, possibly removes damaged or mismatched DNA ends to expose short lengths of single-stranded DNA. In the absence of a sister chromatid, sites of limited homology in the resected region may anneal to begin repair through SSA. Mutation of the Nijmegen breakage syndrome 1 (NBS1) gene results in the radiationsensitive disease Nijmegen breakage syndrome. Affected individuals have immune deficiency, chromosome instability and lymphoreticular tumors.
Defects in DNA repair underlie a number of human genetic diseases that affect a wide variety of body systems but share a constellation of common traits, most notably a predisposition to cancer. These disorders include Ataxia-telangiectasia (AT), a degenerative motor condition caused by failure to repair oxidative damage in the cerebellum, and Xeroderma pigmentosum (XP), a condition characterized by sensitivity to sunlight and linked to a defect in an important ultraviolet (UV) damage repair pathway. In addition, a number of genes that have been implicated in cancer, such as the RAD group, have also been determined to encode proteins critical for DNA damage repair.
Direct Repair:
Also known as reversal of damage can be well demonstrated by two well-known examples - UV-induced DNA dimers by photolyases and the repair of mutagenic methyl lesions by methyltransferases
Photolyases:
Photolyases use the energy in visible light to split ultraviolet (UV)-induced Cyclobutyl dimmers of cytosine and thymine dimers, in a process called photoreactivation. Photolyases work in concert with two molecular cofactors, or chromophores. The first chromophore captures a photon of visible light and the second chromophore uses the energy of the captured photon to initiate an electron transfer that splits the dimer bonds, restoring the dimerized bases to their normal structure. Photolyases have been found in certain bacteria, yeast and marsupials, but they have not been found in mammals. Photoreactivation is absent in Humans. Cyclobutyl dimers in such organisms lacking photoreactivation are repaired by nucleotide Excision repair.
Methyltransferases:
Excision repair:
It is of two types- Base excision repair and Nucleotide excision repair.
Base excision repair:
Base excision repair (BER) is a repair mechanism that aims at complete removal of the damaged base from the DNA by excision and is subsequently replaced by a new one.In addition, BER is a multistep process that involves more than one protein which can be well explained in E. coli. Initially, an enzyme called DNA glycosylase cleaves the glycosydic bond connecting the sugar and the base resulting in removal of the base. As a consequence, an abasic site or a one-base gap is generated in the DNA helix. Another enzyme called AP endonuclease cuts the sugar–phosphate backbone at the abasic site and the hanging remnants are clipped off by a third enzyme, deoxyribophosphodiesterase. Lastly, a DNA polymerase inserts a new nucleotide in the gap and DNA ligase seals the new nucleotide into the DNA. BER can fix a large number of damaged bases with the aid of different glycosylases which are assigned specific roles to remove a few different damaged bases or types of damaged bases. For instance, uracil DNA glycosylases remove uracils that result from damage to cytosines. The 3-methyladenine DNA glycosylases remove alkylated bases, such as 3-methyladenine, which are not repaired by methyltransferases. Yet another glycosylase removes the oxidatively damaged base 8-oxoguanine. In each case, the glycosylase activity results in an abasic site that is repaired by the rest of the BER pathway. Like the DNA-repair methyltransferases, the BER proteins are found in organisms from bacteria to humans.
Nucleotide excision repair:
Nucleotide excision repair is of much broader specificity than base excision repair. The major limitation of BER, in spite of its ability to repair a wide range of altered or adducted bases from DNA is that, it can only be used to repair lesions for which there are specific DNA glycosylases. In order to overcome this drawback where the number of lesion types exceed the number of lesion specific glycosylases, cells employ alternate repair mechanisms. There is another process that can repair many different, nonspecific types of base adducts and the repair process is called Nucleotide excision repair (NER). It deals with extreme forms of dmage like intrastrand crosslinks, modofed bases with large chemical groups. NER identifies the DNA damage by recognizing a distortion in the DNA helix caused by the lesion rather than recognizing specific lesions. For example, the pyrimidine dimers caused by ultraviolet light make a bend, or a kink, in DNA molecules. Crosslinks and bulky adducts like the aflatoxin adduct also distort DNA and are substrates for repair by NER. It repairs cyclobutyl dimmers by a dark process without the reuirement of light energy in organisms which like photoreactivation. Unlike in BER, an important observation to be made in NER is that the process does not involve selective base removal but it eliminates an entire piece of DNA containing the lesion, rather than just the damaged base. This complex, multistep repair mechanism can be best explained in E.coli which requires numerous proteins. The first step is the detection of DNA damage by a complex of two proteins, UvrA and UvrB. Once the damaged site is identified, Uvr A helps Uvr B bind the damaged region tightly, and then UvrA falls off the DNA. A third protein, Uvr C, joins UvrB on the DNA. Uvr B then
cuts the DNA strand at the fifth phosphodiester bond downstream of the damaged site, on one side of the lesion and UvrC cuts the strand on the other side at the eight phosphodiester bond upstream. A fourth protein, called UvrD, then displaces the twelve nucleotide single-stranded piece of DNA containing the lesion, leaving a single-stranded gap in the double-stranded helix. A DNA polymerase then synthesizes a new tract of single-stranded DNA to fill in the gap,and DNA ligase seals the newly made patch into the DNA. This process of repair mentioned above is known as short patch repair process and is absent in eukaryotes but long patch repair process involving 24-29 nucleotides exists.
In higher organisms, like humans, the process of NER appears to be more complicated, involving even more proteins. But the basic pattern of recognition of damage and removal of a segment of DNA containing the damage is the same. Deficiencies in NER proteins in humans cause several severe inherited diseases.
Mismatch repair
Due to the high processivity of DNA polymerases during replication occasional incorporation of an incorrect or mismatched base opposite a normal base is a common phenonmenon. Mismatches as such do not cause DNA damage, in the sense of an altered or damaged base, but they can nevertheless cause mutations. Therefore cells employ mechanisms to inspect the newly formed DNA after replication for mismatches.In E. coli, mismatches in the newly replicated DNA are identified and bound by a dimer of Mut S proteins. With the help of a second protein (Mut L), the Mut S dimer binds and activates a third protein, Mut H. Once it is activated, Mut H can make a nick in the newly synthesized DNA strand containing the mismatched base. Other enzymes then move in and remove a segment of the strand containing the incorrect base. This excision step leaves long gaps of up to several thousand bases, which are repaired by a DNA polymerase and DNA ligase. Like NER and BER, mismatch repair is an excision repair process.
An important feature of mismatch repair is that it is targeted to the newly synthesized DNA strand and not on the original (or template) strand which can cause removal of the correct (template)base, instead of the incorrect (new)one. Cells figure out the new strand based on a chemical modification, called Dam methylation, that is present exclusively on the old strand. The MutH enzyme only cuts newly synthesized DNA strands that are missing this modification.
SOS Response:
SOS repair when genome is extensively damaged. Cells prefer to replicate thedamaged region in a error prone manner rather than dying. It is a coordinated cellular response which aids in the survival of the organisms by effecting the expression of protein that are involved in cell division, replication, recombination and excision repair. About 30 genes are reported to be involved in this process under normal conditions a protein called Lex A repressor binds to SOS box to repress expression of SOS genes. Under conditions of extensive DNA damage or blockage of replication, formation of large single stranded DNA regions occurs, which are recognised by Rec A protein. Single stranded DNA- Rec A complex bind to Lex A and inactivate the repressor which results in the transcription of SOS genes. As a result of this, cell division stops, excision and recombination activities increase. Replication occurs extensively to bypass the damage. SOS response enables the cell either to eliminate the DNA damage or to survive with damaged DNA.
Recombinational Repair:
Recombinational repair is a post replication repair process. The recombinational repair pathway responds to the potentially lethal formation double standed DNA breaks (DSBs) that occur in the genome through the direct action of ionizing radiation, exposure to certain chemicals, or the product of blocked replication forks. There are actually three DSB repair pathways that operate in the cell, and defects in each of these pathways have serious consequences for human health. The mechanism of two of the pathways uses homologous recombination, whereas the other does not. Nonhomologous end rejoining
Nonhomologous end rejoining (NHEJ) is effected without the need for extensive homology between the DNA ends that are to be joined, and it has been considered the principal pathway for DSB repair in mammalian cells. This pathway also processes the site-speci?c DSBs created during variant (diversity) joining (V(D)J) recombination – an essential component for the developmental maturation of the immune system. The mechanism of end rejoining involves the Ku70/80 heterodimer that binds the ends of the double-stranded DNA to be rejoined. This activates the catalytic subunit of DNA-PK, a protein kinase that stabilizes the interaction with the DNA ends. Rejoining is then carried out by the DNA ligase IV/XRCC4 heterodimer.
Homologous recombination and single-strand annealing:
Two pathways use homologous recombination (HR) as a mechanism for DSB repair. HR uses the RAD52 group of proteins to form and resolve Holliday junctions. Mammalian cells carry out HR during mitotic recombination and repair DSBs by HR in late S and G2 phases of the cell cycle, where an undamaged sister chromatid is available. The human breast cancer susceptibility genes breast cancer 1, early onset (BRCA1) and breast cancer 2, early onset (BRCA2) are involved in recombinational repair together with RAD51 homolog (RAD51). In cells with damaged DNA, these three proteins relocate to regions that contain PCNA at sites of DNA damage. ATM, which is mutated in the radiation sensitive disorder ataxia telangiectasia, is proposed to be a damage sensor of DSBs in the HR pathway. ATM phosphorylates BRCA1, which triggers the response to ionizing radiation in mammalian cells.The single-strand annealing (SSA) pathway is a subpathway of HR, which relies on regions of homology to align the strands of DNA to be rejoined. The proteins involved in the pathway are RAD50, MRE11 and NBS1. MRE11, a 30–50 exonuclease, possibly removes damaged or mismatched DNA ends to expose short lengths of single-stranded DNA. In the absence of a sister chromatid, sites of limited homology in the resected region may anneal to begin repair through SSA. Mutation of the Nijmegen breakage syndrome 1 (NBS1) gene results in the radiationsensitive disease Nijmegen breakage syndrome. Affected individuals have immune deficiency, chromosome instability and lymphoreticular tumors.
Defects in DNA repair underlie a number of human genetic diseases that affect a wide variety of body systems but share a constellation of common traits, most notably a predisposition to cancer. These disorders include Ataxia-telangiectasia (AT), a degenerative motor condition caused by failure to repair oxidative damage in the cerebellum, and Xeroderma pigmentosum (XP), a condition characterized by sensitivity to sunlight and linked to a defect in an important ultraviolet (UV) damage repair pathway. In addition, a number of genes that have been implicated in cancer, such as the RAD group, have also been determined to encode proteins critical for DNA damage repair.
Published date : 25 Jun 2014 12:23PM




More Articles


