

Sakshi Education
Cancer is a fatal disease mainly caused by environmental factors that mutate genes encoding critical cell-regulatory proteins. The resultant aberrant cell behaviour leads to expansive masses of abnormal cells that destroy surrounding normal tissue and can spread to vital organs resulting in disseminated disease, it a harbinger of imminent death for patient.
The following closely related terms may be used to designate abnormal growths:
PROPERTIES OF CANCER CELLS
Cancers are classified by the type of cell that resembles the tumourand, therefore, the tissue presumed to be the origin of the tumor. The following general categories are usually accepted:
Benign tumors are named using -oma as a suffix with the organ name as the root. For instance, a benign tumour of the smooth muscle of the uterus is called leiomyoma (the common name of this frequent tumour is fibroid).
Adult cancers
In the USA and other developed countries, cancer is presently responsible for about 25% of all deaths. On a yearly basis, 0.5% of the population is diagnosed with cancer.
Childhood Cancers
Cancer can also occur in young children and adolescents, but it is rare. Some studies have concluded that pediatric cancers, especially leukemia, are on an upward trend.
The age of peak incidence of cancer in children occurs during the first year of life. Leukemia (usually ALL) is the most common infant malignancy (30%), followed by the central nervous system cancers and neuroblastoma. The remainder consists of Wilms' tumor, lymphomas, rhabdomyosarcoma (arising from muscle), retinoblastoma, osteosarcoma and Ewing's sarcoma.
Female and male infants have essentially the same overall cancer incidence rates, but white infants have substantially higher cancer rates than black infants for most cancer types. Relative survival for infants is very good for neuroblastoma, Wilms' tumor and retinoblastoma, and fairly good (80%) for leukemia, but not for most other types of cancer.
CARCINOGEN
They usually function to prevent cell division or to cause death of abnormal cells. Tumour suppressor genes were first discovered by Alfred Knudson through his studies of the inheritance of retinoblastoma, a childhood form of retinal cancer. Mutations that inactivate tumour suppressor genes (called loss-of-function mutations) can be caused by deletion of the tumour suppressor gene, by a pointmutation that leads to a functionally inactive tumour suppressor gene-encoded protein, or by a mutation that disrupts the promoter region of the tumour suppressor gene, leading to a loss or reduction in its expression. In most cases, tumour suppressor gene loss-of-function mutations are recessive, meaning that both copies of a cell's tumour suppressor gene must be mutated in order for cancer to occur. The 2 most important are p53 and Rb.
PROTO-ONCOGENE-A cellular (host) gene that is homologous with a similar gene (v-onc) that is found in a transforming virus. They are given the names of form c-onc.
Oncogenes are genes whose products have the ability to transform eukaryotic cells so that they grow in a manner analogous to tumour cells. oncogenes carried by retroviruses have names of the form v-onc.A cellular oncogene can only induce transformation after mutation or some other change in the cell’s genome. They result in dominant gain-of-function mutations.Oncogene mutations act dominantly
Proteins encoded by proto-oncogenes are .
First recognised oncogene was v-src, identified in Rous sarcoma virus. The viral oncogene causes cells to be released from their normal growth controls. cDNA probes against viral oncogenes show similar homologues in NORMAL eucaryotic cells. These cellular homologues are not identical to the viral oncogene and it seems that the virus has captured a cellular gene during its evolution. DNA containing oncovirus.
Most somatic cells have a ‘molecular clock’ that limits the number of times they can divide. This is known as the ‘Hayflick limit’, and in most cells this is between 50 and 70 doublings, after which cells enter a state of senescence and cease dividing. The molecular clock is telomere shortening. Telomeres are protective caps on the ends of chromosomes, commonly composed of short, tandemly repeated, sequences that are guanosine-rich (e.g. (GGGTTA)n).
A special enzymecalled telomerase acts on telomeres to extend their length, but this enzyme is notactive in most normal cells. Therefore, most normal cells can only divide a certainnumber of times before their telomeres shorten to the extent that critical genetic information is lost. Cancer cells are immortalized cells and, althoughthe cell of origin of some cancers may have sufficienttelomerase activity to prevent significant telomere erosion, most cancers probably originate in a telomerase-negativecell but they escape eventual cellular death by reactivationof telomerase.
APOPTOSIS
Net tumour growth is due to the cell production rate through mitosis exceeding the cell loss rate through cell death. When DNA is damaged, signals go to both repair and apoptotic pathways, and if repair cannot be effected then the cell undergoes apoptosis’. In response to damage, normal cells upregulate p53 which acts as a transcription factor for cell cycle arrest and apoptosis, p53-mutant cells cannot carry out this protective arrest or apoptosis and survive with what otherwise would be lethal genetic damage, perhaps explaining why p53 mutations are so common in human cancers.
CELL ADHESION
Changes in expression of cell adhesion molecules (CAMs) are crucial to many aspects of tumour behaviour. In primary tumours, downregulation of the type IV collagen and laminin receptors leads to loss of cell attachment from the basement membrane and isimportant for invasion. Conversely, expression of particular integrins may be crucial for metastasis. Similarly, upregulationof integrins on tumour cells facilitates adhesion toendothelium .Reduced expression of E-cadherin, facilitates cell detachment from the primary tumour mass, invasion and metastasis
CELL SIGNALLING
Autocrine signalling is generally found in cancer cells, which allows them to grow autonomously in culture devoid of growth factors, and bestows upon them some independence from normal growth restraints. Apart from polypeptides, lipophilic hormones such as steroids, retinoids and thyroid hormones are potent regulators of cell behaviour, and many cancers of their target tissues are hormone-dependent and thus are responsive to hormone ablation therapy. In the prevention or treatment of breast cancer, steroid hormone analogues such as tamoxifen are used tomimic the action of the natural oestrogen, eliciting a muchweaker oestrogenic response
CELL CYCLE REGULATION
Growth factor binding leads to receptor dimerization and phosphorylation, activation of Ras and the mitogenactivatedprotein kinase (MAPK) signal transduction pathway leading to cyclin D production. Many of the genes encoding growth factors, receptors, components of the signal transduction pathway and cyclins are proto-oncogenes, genes that whenactivated by mutation can contribute to cancer development. pRb, p53 and the cyclin-dependent kinase inhibitors (CKIs) all act as a brake on cell cycling and are the products of tumour suppressor genes (TSGs); when inactivated by mutation, loss or viral proteins, they also contribute to cancer development.
The phosphorylation of pRb is necessary for the release of E2F–DP dimers that promote the transcription of cell cycle-associated genes. pRb can be inactivated by virally encoded oncoproteins such as adenovirus E1a and human papillomavirus (HPV) E7. p53 is negatively regulated by Mdm2, an enzyme required to produce a polyubiquitinated p53 for degradation by the proteasome. p53 can be disabled by adenovirus E1b and HPV E6. The Ink4a locus also encodes p14ARF whose function is to activate p53 by binding to and inactivating Mdm2, making ARF another TSG. DNA, deoxyribonucleic acid; DHFR, dihydrofolatereductase;TGFb, transforming growth factor b.
DNA REPAIR AND GENETIC INSTABILITY
Most cancers arise due to alterations in genes involved in DNA replication, repair, telomere stabilization and chromosome segregation, and could lead to point mutations, deletions or additions of a few nucleotides, translocations, and even losses or gains of whole or parts of chromosomes
Cells from patients with ataxia telangiectasia cannot effect cell cycle arrest after irradiation-induced DNA damage, referred to as radiation-resistant DNA synthesis. Patients with xerodermapigmentosum suffer from a defect in nucleotide excision repair, becoming highly sensitive to ultraviolet light-induced damage with a 2000-fold increased risk of developing skin cancer
ANGIOGENESIS-
It is the formation of new blood vessels from existing vessels. ‘Stressed’ tumour cells, perhaps suffering from hypoxia, release proangiogenic growth factors like VEGF, FGF that, in concert with growth factors produced by the endothelial cells stimulate endothelial cell migration and division. The stimulated endothelial cells release extracellular matrix (ECM)-busting enzymes such as urokinase-type and tissue-type plasminogen activators, and collagenases, as well as inhibitors such as plasminogen activator inhibitor 1. Endothelial cells also release basement membrane components such as laminin, type IV collagen andtenascin, and express ECM receptors such as the a5b3 and a5b5 integrins which help inmigration and proliferation. New matrix is formed and it stabilised by pericytes.
Antiangiogenictherapy has minimal side effects because angiogenesis is a physiological host response. Pharmacological blockade should not lead to thedevelopment of resistance since normal endothelial cells lack the genetic instability of cancer cells that is responsible for the emergence of drug-resistant clones. As each capillary in a tumour supplies many hundreds of tumour cells, targeting the endothelium will lead to a potentiation of the antitumour effect. Therapeutic agents have direct access to the endothelium. The action of inhibitors ranges from blocking endothelial proliferation, antagonizing growth factor receptors, suppressing proteolytic enzyme secretion, to blocking integrin expression so making cells marooned from the ECM and consequently undergoing apoptosis.
METASTASIS
If the tumour is amenable to surgery, then surgery is the single most effective tool in its cure.
Targeted radiotherapy is another option, as are combinations of anticancer drugs.
Most conventional anticancer drugs have been designed with deoxyribonucleic acid (DNA) synthesis as their target. Therein lies the problem, in that tumour cells are not the only proliferating cells in the body; cells that line the alimentary tract, bone marrow cells that generate red blood cells and cells to fight infection, and epidermal cells including those that generate hair are all highly proliferative. Thus, patients with cancer receiving chemotherapy commonly suffer unwanted (hair loss) and sometimes potentially life-threatening (anaemia and proneness to infections) side effects that limit treatment.
The new generation of drugs affect the signals that promote or regulate the cell cycle, growth factors and their receptors, signal transduction pathways and pathways affecting DNA repair and apoptosis. Each of these pathways may be affected by activating mutations that predispose to cancer and, thus, offer the potential as a target for inhibition. Other strategies focus on either attempting to target tumour cells specifically by conjugating cell toxins to tumour-specific antibodies (magicbullets), or slowing down cancer progression by affecting cell adhesion, proteolytic enzyme activity and angiogenesis.
REFERENCES
The following closely related terms may be used to designate abnormal growths:
- Neoplasia and Neoplasm are the scientific designations for cancerous diseases. This group contains a large number of different diseases. Neoplasms can be benign or malignant.
- Cancer is a widely used word that is usually understood as synonymous with malignant neoplasm. It is occasionally used instead of carcinoma, a sub-group of malignant neoplasms. Because of its overwhelming popularity relative to 'neoplasia', it is used frequently instead of 'neoplasia', even by scientists and physicians, especially when discussing neoplastic diseases as a group.
- Tumour in medical language simply means either swelling, or lump, or neoplastic or inflammatory or other. In common language, however, it is synonymous with 'neoplasm', either benign or malignant. This is inaccurate since some neoplasms do not usually form tumors, for example leukemia or carcinoma in situ.
- Paraneoplasia is a disturbance associated with a neoplasm but not related to the invasion of the primary or a secondary (metastatic) tumour. Disturbances can be hormonal, neurological, hematological, biochemical or otherwise clinical.
- These tumors remain clustered together in a single cell mass and do not spread to other sites. The surface interaction molecules holding the tissue together keep the tumourcells localized e.g. benign tumourcells of liver stay in liver.
- A fibrous capsule usually delineates the extent of benign tumor. They become serious problem only when their sheer bulk interferes with normal function or if they secret excess amount of hormones.
- Examples are warts.
- Most of the tumours do not remain in their original site instead they metastasize. Invasiveness and spread is the major characteristic that differentiate the benign tumors from malignant.
- They express proteins characteristics of the cell type from which it arose and grow and divide rapidly than normal.
PROPERTIES OF CANCER CELLS
- Altered morphology (rounded shape, refractile in phase contrast microscope)
- Loss of contact inhibition i.e. ability to grow over one another
- Anchorage independence-ability to grow without attachment to solid substrate
- Immortalization-ability to proliferate indefinitely
- Reduced requirement for mitogenic growth factors
- High saturation density –ability to accumulate large number of cells in culture dish
- Inability to halt proliferation in response to deprivation of growth factors
- Increased transport of glucose
- Tumorigenicity
Cancers are classified by the type of cell that resembles the tumourand, therefore, the tissue presumed to be the origin of the tumor. The following general categories are usually accepted:
- Carcinoma: malignant tumours derived from epithelial cells. This group represents the most common cancers, including the common forms of breast, prostate, lung and colon cancer.
- Lymphoma and Leukemia: malignant tumours derived from blood and bone marrow cells.
- Sarcoma: malignant tumours derived from connective tissue, or mesenchymal cells.
- Mesothelioma: tumours derived from the mesothelial cells lining the peritoneum and the pleura.
- Glioma: tumours derived from glia, the most common type of brain cell.
- Germinoma: tumours derived from germ cells, normally found in the testicle and ovary.
- Choriocarcinoma: malignant tumours derived from the placenta.
Benign tumors are named using -oma as a suffix with the organ name as the root. For instance, a benign tumour of the smooth muscle of the uterus is called leiomyoma (the common name of this frequent tumour is fibroid).
Adult cancers
In the USA and other developed countries, cancer is presently responsible for about 25% of all deaths. On a yearly basis, 0.5% of the population is diagnosed with cancer.
Childhood Cancers
Cancer can also occur in young children and adolescents, but it is rare. Some studies have concluded that pediatric cancers, especially leukemia, are on an upward trend.
The age of peak incidence of cancer in children occurs during the first year of life. Leukemia (usually ALL) is the most common infant malignancy (30%), followed by the central nervous system cancers and neuroblastoma. The remainder consists of Wilms' tumor, lymphomas, rhabdomyosarcoma (arising from muscle), retinoblastoma, osteosarcoma and Ewing's sarcoma.
Female and male infants have essentially the same overall cancer incidence rates, but white infants have substantially higher cancer rates than black infants for most cancer types. Relative survival for infants is very good for neuroblastoma, Wilms' tumor and retinoblastoma, and fairly good (80%) for leukemia, but not for most other types of cancer.
CARCINOGEN
- A carcinogen is any substance or agent that, because of the way it affects cell DNA, can cause cancer.
- Some initiate and some promote cancer implying the existence of different stages of cancer, which may cause epigenetic changes or act directly.
- Carcinogens may be
- chemical substances;
- physical agents, such as asbestos dust;
- or biological agents, such as certain viruses and bacteria
- In the workplace, carcinogenic substances may be inhaled, absorbed through the skin or even ingested in some cases.
- Exogenous carcinogens
- Chemical carcinogens
- Direct acting - alkylating, acylating agents
- Procarcinogens - that require metabolic activation. Eg;Benz(a) anthracene and Benzo(a) pyrene.
- Natural plant and animal products- aflatoxins, mycotoxinsetc are potent liver carcinogens
- Physical carcinogens-radiations, heat (Kangri cancer Kashmir)
- Viruses
- Hormones
- Chemical carcinogens
- Endogenous Carcinogens
- Arise due to errors in metabolism of hormones or bile acids.
-
- Mutations causing cancer occur mostly in somatic cells and not in germ line cells
- Somatic cell mutations are not passed on to next generation
- Susceptibility to cause cancer is inherited in some cancers
- The multi-hit model, which proposes that multiple mutations are needed to cause cancer, is consistent with the genetic homogeneity of cells from a given tumor, the observed increase in the incidence of human cancers with advancing age, and the cooperative effect of oncogenic transgenes on tumourformation in mice.
- Colon cancer develops through distinct morphological stages that commonly are associated with mutations in specific tumor-suppressor genes and proto-oncogenes
- Cancer forming process is called oncogenesis
- It is interplay between genetics and environment
- Most cancer arise due to mutations in genes.When a mutation occurs in a somatic cell it gives rise to a daughter cell giving a clone of altered cells
- Rarely a mutation in single gene will lead to onset of cancer.A series of mutations in multiple genes creates a progressively more rapidly proliferating cell type that escapes normal growth restraints creating an opportunity for more mutations
- The clone of cells grows in a tumour and it can be both monoclonal as well as polyclonal.
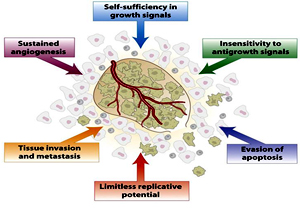
- the ability to divide in the absence of growth factor stimulation,
- the ability todivide in the presence of anti-growth signals,
- the inability to undergo apoptosis,
- the ability to maintain telomere length despite repeated cell divisions,
- stimulation ofangiogenesis, and
- the ability to invade surrounding tissues and metastasize toother parts of the body
They usually function to prevent cell division or to cause death of abnormal cells. Tumour suppressor genes were first discovered by Alfred Knudson through his studies of the inheritance of retinoblastoma, a childhood form of retinal cancer. Mutations that inactivate tumour suppressor genes (called loss-of-function mutations) can be caused by deletion of the tumour suppressor gene, by a pointmutation that leads to a functionally inactive tumour suppressor gene-encoded protein, or by a mutation that disrupts the promoter region of the tumour suppressor gene, leading to a loss or reduction in its expression. In most cases, tumour suppressor gene loss-of-function mutations are recessive, meaning that both copies of a cell's tumour suppressor gene must be mutated in order for cancer to occur. The 2 most important are p53 and Rb.
PROTO-ONCOGENE-A cellular (host) gene that is homologous with a similar gene (v-onc) that is found in a transforming virus. They are given the names of form c-onc.
Oncogenes are genes whose products have the ability to transform eukaryotic cells so that they grow in a manner analogous to tumour cells. oncogenes carried by retroviruses have names of the form v-onc.A cellular oncogene can only induce transformation after mutation or some other change in the cell’s genome. They result in dominant gain-of-function mutations.Oncogene mutations act dominantly
Proteins encoded by proto-oncogenes are .
- Positive acting growth factors–PDGF [sis gene product]
- Growth factor receptor-EGF-R [erb-B gene product]
- Non-Receptor protein tyrosine kinase-Src[srcgene product]
- Transcription factor-c-Myc
- Signal transduction proteins-like G proteins-Ras [ras gene product]
- Cell cycle control proteins-cyclin D1
- Apoptosis related genes-bcl-2
- Activating mutation of one of the two alleles of proto-oncogenes convert it to oncogene which can induce transformation in cultured cells and cancer in animals
- Activation of proto-oncogenes into an oncogene can occur by
- Point mutation
- Gene amplification and
- Gene/chromosomal translocation-eg-The translocation of the tip of chromosome 9 to the tip of chromosome 22 forms an abnormal chromosome known as the Philadelphia chromosome. Hematopoietic cells with this translocation give rise to chronic myelogenous leukemia.
First recognised oncogene was v-src, identified in Rous sarcoma virus. The viral oncogene causes cells to be released from their normal growth controls. cDNA probes against viral oncogenes show similar homologues in NORMAL eucaryotic cells. These cellular homologues are not identical to the viral oncogene and it seems that the virus has captured a cellular gene during its evolution. DNA containing oncovirus.
- DNA tumour viruses have transforming genes which are their own (viral oncogenes) ,they do not carry cellular oncogenes like RNA viruses
- DNA virus oncogenes act by inactivating host tumour suppressor gene products (p53, pRB) or they influence the cell cycle and apoptosis Eg: Hepatatis B virus (liver cancer)
Epstein Barr virus (Burkitt’s lymphoma)
Human papilloma Virus (cervical cancer)
RNA containing oncovirus.
Eg: rous sarcoma virus- v-src
Simian sarcoma virus– v-sis
Most somatic cells have a ‘molecular clock’ that limits the number of times they can divide. This is known as the ‘Hayflick limit’, and in most cells this is between 50 and 70 doublings, after which cells enter a state of senescence and cease dividing. The molecular clock is telomere shortening. Telomeres are protective caps on the ends of chromosomes, commonly composed of short, tandemly repeated, sequences that are guanosine-rich (e.g. (GGGTTA)n).
A special enzymecalled telomerase acts on telomeres to extend their length, but this enzyme is notactive in most normal cells. Therefore, most normal cells can only divide a certainnumber of times before their telomeres shorten to the extent that critical genetic information is lost. Cancer cells are immortalized cells and, althoughthe cell of origin of some cancers may have sufficienttelomerase activity to prevent significant telomere erosion, most cancers probably originate in a telomerase-negativecell but they escape eventual cellular death by reactivationof telomerase.
APOPTOSIS
Net tumour growth is due to the cell production rate through mitosis exceeding the cell loss rate through cell death. When DNA is damaged, signals go to both repair and apoptotic pathways, and if repair cannot be effected then the cell undergoes apoptosis’. In response to damage, normal cells upregulate p53 which acts as a transcription factor for cell cycle arrest and apoptosis, p53-mutant cells cannot carry out this protective arrest or apoptosis and survive with what otherwise would be lethal genetic damage, perhaps explaining why p53 mutations are so common in human cancers.
CELL ADHESION
Changes in expression of cell adhesion molecules (CAMs) are crucial to many aspects of tumour behaviour. In primary tumours, downregulation of the type IV collagen and laminin receptors leads to loss of cell attachment from the basement membrane and isimportant for invasion. Conversely, expression of particular integrins may be crucial for metastasis. Similarly, upregulationof integrins on tumour cells facilitates adhesion toendothelium .Reduced expression of E-cadherin, facilitates cell detachment from the primary tumour mass, invasion and metastasis
CELL SIGNALLING
Autocrine signalling is generally found in cancer cells, which allows them to grow autonomously in culture devoid of growth factors, and bestows upon them some independence from normal growth restraints. Apart from polypeptides, lipophilic hormones such as steroids, retinoids and thyroid hormones are potent regulators of cell behaviour, and many cancers of their target tissues are hormone-dependent and thus are responsive to hormone ablation therapy. In the prevention or treatment of breast cancer, steroid hormone analogues such as tamoxifen are used tomimic the action of the natural oestrogen, eliciting a muchweaker oestrogenic response
CELL CYCLE REGULATION
Growth factor binding leads to receptor dimerization and phosphorylation, activation of Ras and the mitogenactivatedprotein kinase (MAPK) signal transduction pathway leading to cyclin D production. Many of the genes encoding growth factors, receptors, components of the signal transduction pathway and cyclins are proto-oncogenes, genes that whenactivated by mutation can contribute to cancer development. pRb, p53 and the cyclin-dependent kinase inhibitors (CKIs) all act as a brake on cell cycling and are the products of tumour suppressor genes (TSGs); when inactivated by mutation, loss or viral proteins, they also contribute to cancer development.
The phosphorylation of pRb is necessary for the release of E2F–DP dimers that promote the transcription of cell cycle-associated genes. pRb can be inactivated by virally encoded oncoproteins such as adenovirus E1a and human papillomavirus (HPV) E7. p53 is negatively regulated by Mdm2, an enzyme required to produce a polyubiquitinated p53 for degradation by the proteasome. p53 can be disabled by adenovirus E1b and HPV E6. The Ink4a locus also encodes p14ARF whose function is to activate p53 by binding to and inactivating Mdm2, making ARF another TSG. DNA, deoxyribonucleic acid; DHFR, dihydrofolatereductase;TGFb, transforming growth factor b.
DNA REPAIR AND GENETIC INSTABILITY
Most cancers arise due to alterations in genes involved in DNA replication, repair, telomere stabilization and chromosome segregation, and could lead to point mutations, deletions or additions of a few nucleotides, translocations, and even losses or gains of whole or parts of chromosomes
Cells from patients with ataxia telangiectasia cannot effect cell cycle arrest after irradiation-induced DNA damage, referred to as radiation-resistant DNA synthesis. Patients with xerodermapigmentosum suffer from a defect in nucleotide excision repair, becoming highly sensitive to ultraviolet light-induced damage with a 2000-fold increased risk of developing skin cancer
ANGIOGENESIS-
It is the formation of new blood vessels from existing vessels. ‘Stressed’ tumour cells, perhaps suffering from hypoxia, release proangiogenic growth factors like VEGF, FGF that, in concert with growth factors produced by the endothelial cells stimulate endothelial cell migration and division. The stimulated endothelial cells release extracellular matrix (ECM)-busting enzymes such as urokinase-type and tissue-type plasminogen activators, and collagenases, as well as inhibitors such as plasminogen activator inhibitor 1. Endothelial cells also release basement membrane components such as laminin, type IV collagen andtenascin, and express ECM receptors such as the a5b3 and a5b5 integrins which help inmigration and proliferation. New matrix is formed and it stabilised by pericytes.
Antiangiogenictherapy has minimal side effects because angiogenesis is a physiological host response. Pharmacological blockade should not lead to thedevelopment of resistance since normal endothelial cells lack the genetic instability of cancer cells that is responsible for the emergence of drug-resistant clones. As each capillary in a tumour supplies many hundreds of tumour cells, targeting the endothelium will lead to a potentiation of the antitumour effect. Therapeutic agents have direct access to the endothelium. The action of inhibitors ranges from blocking endothelial proliferation, antagonizing growth factor receptors, suppressing proteolytic enzyme secretion, to blocking integrin expression so making cells marooned from the ECM and consequently undergoing apoptosis.
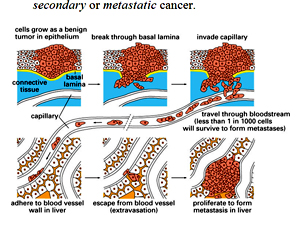
METASTASIS
- er cells have the ability to spread, they can spread through the lymphatic system or bloodstream to distant organs such as the bone, lung, and liver.
- When a cancer spreads, it retains the properties of the original cancer.
- The original cancer is called the primary cancer. A cancer that has spread to another site is called a secondary or metastatic cancer.
If the tumour is amenable to surgery, then surgery is the single most effective tool in its cure.
Targeted radiotherapy is another option, as are combinations of anticancer drugs.
Most conventional anticancer drugs have been designed with deoxyribonucleic acid (DNA) synthesis as their target. Therein lies the problem, in that tumour cells are not the only proliferating cells in the body; cells that line the alimentary tract, bone marrow cells that generate red blood cells and cells to fight infection, and epidermal cells including those that generate hair are all highly proliferative. Thus, patients with cancer receiving chemotherapy commonly suffer unwanted (hair loss) and sometimes potentially life-threatening (anaemia and proneness to infections) side effects that limit treatment.
The new generation of drugs affect the signals that promote or regulate the cell cycle, growth factors and their receptors, signal transduction pathways and pathways affecting DNA repair and apoptosis. Each of these pathways may be affected by activating mutations that predispose to cancer and, thus, offer the potential as a target for inhibition. Other strategies focus on either attempting to target tumour cells specifically by conjugating cell toxins to tumour-specific antibodies (magicbullets), or slowing down cancer progression by affecting cell adhesion, proteolytic enzyme activity and angiogenesis.
REFERENCES
- Chial, H. (2008) Genetic regulation of Cancer. Nature Education 1(1):67
- Hanahan, D., & Weinberg, R. A. (2000) The Hallmarks of Cancer. Cell 100, 57–70.
- Christofi G and Semb H (1999) The Role of the Cell Adhesion Molecule E-Cdherin as a Tumour Suppressor Gene. Trends in Biochemical sciences 24, 73-76.
- Greider CW (1999) Telomerase Activation, Ones Step on the Road to Cancer. Trends in Genetics. 15,109-112.
- Langauer C, Kinzler KW and Vogelstein B (1999) Genetic instabilities in Human cancer. Nature, 396, 643-649
Published date : 14 May 2014 05:39PM




More Articles


