

Sakshi Education
Primary Structure
The primary level of structure refers to the linear sequence of amino acids along a polypeptide chain and the location of covalent bonds, namely disulfide bonds, between chains or within a chain. Proteins can be identified explicitly based on the primary structure. In addition the chemical and biological characteristics of a protein are determined by the primary structure and it also specifies the higher levels of protein structure
Formation of the Polypeptide Chain
Amino acids are joined end to- end during protein synthesis through covalent bonds. The a-carboxyl group of the first amino acid is linked to the a-amino group of the next amino acid to generate a peptide bond and eliminate a water molecule. In this process the amino group of the first amino acid of a polypeptide chain and the carboxyl group of the last amino acid remain intact.
Therefore a polypeptide chain has two different ends, names N-terminus and C-terminus, respectively. The process of polymerization of Amino acids occurs from N-terminus to the C-terminus and is also denoted with numbers in the same direction. For example Lys48 represents a Lysine at position 18 from the amino terminus and the amino acid sequence of a polypeptide is generally written from left to right.
A polypeptide chain can be divided into two parts- the ‘main chain’ or ‘backbone’ and a variable part. The main chain comprises of a regularly repeating part forming the backbone from which the variable part arises. A variable part consists of different side chains. An amino acid unit when incorporated into a polypeptide chain is called an amino acid ‘residue’ and differs from the corresponding amino acid by lacking a water molecule. Specific names of amino acid residues are derived from the corresponding free amino acid, e.g. arginyl residue (arginine), lysyl residue (lysine) and so on.
The peptide bond –CO–NH– between two successive residues is a relatively rigid planar structure because of its partial double-bond character due to resonance. As a consequence, rotation of this bond is restricted. However, there is rotational freedom about the single bonds that link each Ca atom to the N and C atoms of peptide bonds. The angle of rotation between the N–Ca bonds is denoted as phi (f) and the angle around the Ca –C bond from the Ca –atom is denoted as psi (?). The planar peptide bond can theoretically exist in two different configurations, the trans and cis forms. The trans form is energetically favoured and is thus more stable over the cis form.
Determination of the Primary Structure
Insulin, a protein hormone of 51 residues was the first protein whose complete amino acid sequence was determined in 1953 by Frederick Sanger. The first step in determining the sequence of an amino acid is to identify the N-terminal and C-terminal residues. Chemical compounds like 1-fluoro-2, 4-dinitrobenzene or dansyl chloride or cyanate are used to label and thereby identify the N terminal residue. Likewise, the C-terminal residue can be identified by converting the a-carboxyl groups of peptide bonds into hydrazides by hydrazinolysis. Enzymatic methods causing sequential release of single amino acids from the ends of chains using specific exopeptidases, namely aminopeptidase and carboxypeptidase can also be employed.
Edman degradation is one of the most efficient procedures for determining the amino acid sequence of a protein, in which amino acids are removed sequentially, one at a time, from the N-terminus without affecting the other regions of the protein. The terminal a-amino group is first reacted with phenylisothiocyanate in alkaline conditions to yield the phenylthiocarbamoyl derivative. The derivative is then released from the rest of the chain, by acid treatment, in the form of a cyclic compound that rearranges in aqueous solution to the phenylthiohydantoin. Chromatographic techniques can be used to identify the phenylthiohydantoin derivative. The procedure can be repeated as many times as necessary to determine the complete amino acid sequence of the protein. Such recurrent analysis is usually performed in an automatic amino acid sequencer.
In case of larger proteins, the protein is initially fragmented, followed by sequencing by the Edman method. In order to execute such fragmentation in specific manner chemicals like Cyanogen bromide, hydroxylamine and N-chlorosuccinimide can be used. Alternatively enzymes like trypsin, pepsin, elastase and thermolysin can be employed. Additionally, techniques like Mass spectrometry can also be used to determining the primary structure of proteins. The technique relies on making the protein volatile by chemical treatment of side-chains and then fragmented nonspecifically with an electron beam. The protein fragments are separated according to their mass-to-charge ratio and identified. Ionisation of proteins with a high-energy beam of atoms or ions can be employed to prevent chemical modification of the protein.
Covalent Modifications of Proteins
Proteins undergo several covalent modifications during or after their biosynthesis. Proteolytic cleavage of the polypeptide chain is one such instance which involves removal of one or several residues from the polypeptide. Such modifications which attain a large biological significance are very helpful in removal of signal peptides and converting inactive form of proteins like zymogens, prohormones, proproteins and preproproteins into active forms.
Protein splicing is another mechanism where excision of internal segments (inteins) of a polypeptide occurs.
So far, about 200 modifications have been detected which can be either reversible or irreversible. The major modifications of the proteins include the addition of lipds, carbohydrates, phosphates and hydroxyl groups etc.
Addition of lipid polar groups like myristoyl groups occurs at the N- terminus and glycosyl-phosphatidylinositol and farnesyl groups at their C- terminus of proteins. Glycosylation is of two types- N-linked glycosylation and O- linked glycosylation.
The N-type occurs on the nitrogen atom of asparaginyl side-chains and O-type, on the oxygen atoms of hydroxyls, particularly those of seryl and threonyl residues.
Another key modification that specifically occurs at the hydroxyl groups is Phosphorylation. It mainly targets the hydroxyl groups of seryl, threonyl and tyrosyl residues in a wide array of proteins belonging to all biological systems and also at aspartyl and histidyl residues in the particular case of bacterial proteins. Hydroxylation is mainly targeted at prolyl and lysyl residues and is highly essential for the maturation and secretion of proteins such as collagens. Other modifications include methylation, acetylation, sulfation, Carboxylation, adenosine diphophste (ADP)-ribosylation, nucleotidylation and amidation.
Addition of non-peptidic prosthetic groups, either covalently or noncovalently like haems, porphyrins, nucleotides and metal ions is essential for some proteins like heteroproteins.
Secondary Structure
Secondary structures are the structures, which are regularly repeating local structures stabilized by hydrogen bonds. The most common examples are the alpha helix and sheet. Because secondary structures are local, many regions of different secondary structure can be present in the same protein molecule.
Prediction of Protein Secondary Structure
Hydrogen bonding interactions between the electronegative carbonyl oxygen atoms and the electropositive amide hydrogen atoms in the backbone chain of the molecule determine the secondary structures that can be adopted by polypeptides in proteins. Although several secondary structures with reasonable hydrogen bonding networks are possible theoretically, only a few possibilities are observed in polypeptides composed of L-amino acids (proteins) due to the limitation on the configuration of the backbone of each amino acid residue.
The Ramachandran Angles
Each amino acid residue had two bonds that can rotate freely generating angles that define the conformation of that residue in a protein and are called the Ramachandran angles, ? (psi) and f (phi).
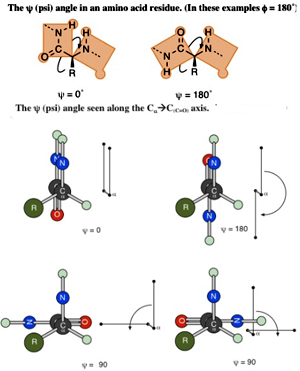
The ? (psi) Angle
The bond from the a-carbon to the carbonyl group (at the C-terminus) of the amino acid residue can rotate and turn the whole plane of the amide group, which includes the carbonyl carbon, in a 360-degree range. This angle is measured by looking along that bond with the carbon of the carbonyl group in the rear and the a-carbon to the front.
The apparent angle measured between the two bonds to nitrogen can be seen as coming out of the axis of the CaC(C=O) bond. This angle is labeled ? (psi) and is measured from–180° to +180° with the positive direction being when the rear group is turned in the clockwise direction so that the rear nitrogen bond is clockwise of the front nitrogen bond (or when the front group is turned in counterclockwise direction so that the rear nitrogen bond is clockwise of the front).
The f (phi) Angle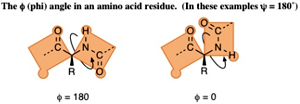 The bond from the nitrogen (at the N-terminus) to the a-carbon of the amino acid residue can rotate and turn the whole plane of the other amide group, which includes the nitrogen, in a 360-degree range.
The bond from the nitrogen (at the N-terminus) to the a-carbon of the amino acid residue can rotate and turn the whole plane of the other amide group, which includes the nitrogen, in a 360-degree range.
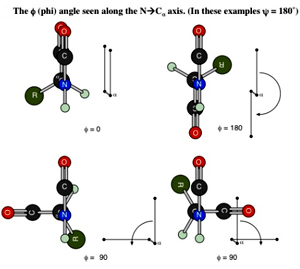 This angle is measured by looking along that bond with the nitrogen atom in front and the a-carbon to the rear. The apparent angle measured between the two bonds to the carbonyl carbons that can be seen as coming out of the axis of the N-Ca bond. This angle is labeled f (phi) and is measured from –180° to +180° with the positive direction being the rear group is turned in clockwise direction so that the rear carbonyl bond is clockwise of the front carbonyl bond (or when the front group is turned in counterclockwise direction so that the rear carbonyl bond is clockwise of the front).
This angle is measured by looking along that bond with the nitrogen atom in front and the a-carbon to the rear. The apparent angle measured between the two bonds to the carbonyl carbons that can be seen as coming out of the axis of the N-Ca bond. This angle is labeled f (phi) and is measured from –180° to +180° with the positive direction being the rear group is turned in clockwise direction so that the rear carbonyl bond is clockwise of the front carbonyl bond (or when the front group is turned in counterclockwise direction so that the rear carbonyl bond is clockwise of the front).
The Ramachandran Plot
The ? and from f can be varied from –180° to 180° theoretically (that is 360° of rotation for each) but many combinations of these angles are almost never observed and some are very, very common in proteins. The Ramachandran plot is a plot of the torsional angles - phi (f)and psi (?) - of the residues (amino acids) contained in a peptide by plotting the f values on the x-axis and the ? values on the y-axis.Plotting the torsional angles in this way graphically shows which combination of angles are possible.
Steric Limits of ? and f
A space occupied by an atom cannot be occupied by another atom at the same time. Atoms are connected by Covalent bonds which cannot be broken but allow rotations. As mentioned earlier there are only two angles that rotate in a given residue, ? and f. But not all combinations are possible due to physical clashes of atoms in 3-dimensional space. These physical clashes are called steric interactions and the limit the available values for ? and f
The Allowed Regions in a Ramachandran Plot
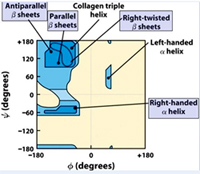 The ß-sheet structures feature ? and f angle combinations of phi = -110 to -140 and psi = +110 to +135 that appear in the upper left corner of the plot. The a-helix regions have ? and f angle combinations that appear in the lower left quadrant with repeating values of phi ~-57o and psi ~-47o give a right-handed helical fold (the alpha-helix) and with values of phi=49 and psi = 26 gives a left handed a-helix.
The ß-sheet structures feature ? and f angle combinations of phi = -110 to -140 and psi = +110 to +135 that appear in the upper left corner of the plot. The a-helix regions have ? and f angle combinations that appear in the lower left quadrant with repeating values of phi ~-57o and psi ~-47o give a right-handed helical fold (the alpha-helix) and with values of phi=49 and psi = 26 gives a left handed a-helix.
The right-handed a-helix is much more stable and is much more common in proteins. Collagen triple helix shows a phi= -51 and psi =153.These structures have ? and f angle combinations that avoid serious steric clashes between the atoms of the amide backbone and side chain groups. Glycine residues do not have the steric constraints of the other 19 residues. Proline residues are conformationally restricted due to the ring being part of the backbone.
Alpha Helix: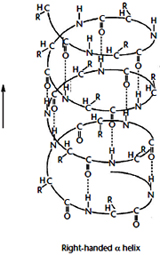 The a-helix structure was first predicted by Linus Pauling and Robert Corey in 1951 on the basis of crystallographic analyses of the structures of various small molecules. The a-helix is a clockwise turning or right-handed rod like structure. Right handed helix can be explained as turning in the direction of the fingers of the right hand when the thumb indicates the line of sight. Its inner part is formed by the coiled polypeptide main chain and the surface by the side-chains projecting outwards in a helical arrangement.
The a-helix structure was first predicted by Linus Pauling and Robert Corey in 1951 on the basis of crystallographic analyses of the structures of various small molecules. The a-helix is a clockwise turning or right-handed rod like structure. Right handed helix can be explained as turning in the direction of the fingers of the right hand when the thumb indicates the line of sight. Its inner part is formed by the coiled polypeptide main chain and the surface by the side-chains projecting outwards in a helical arrangement.
A single turn of the helix contains 3.6 residues and, since a residue extends 1.5A °, the pitch of the helix is 5.4A °(1.5×3.6). It is stabilized by hydrogen bonds between the C=O group of each amino acid residue of the main chain and the N–H group of the residue located four residues away in the amino acid sequence This kind of repeated hydrogen bonding and repeating torsion angle values of phi ~-57o and psi ~-47o defines an a-helix. Orientation of all the hydrogen bonds and peptide groups is in the same direction, nearly parallel to the helix axis. Since each peptide bond possesses an individual dipole moment, the overall effect is a cumulative macrodipole for the helix with a positive charge at the amino end and a negative charge at the carboxyl end.
A left-handed a-helix is also possible sterically, but the side-chains are too close to the main chain and, therefore, this conformation is unstable and rarely encountered in natural polypeptides. Other types of a helix can be predict with hydrogen bonds between residues nearer together (n+3) or farther apart (n+5). The former is called the 310 helix ( hydrogen bonding) and the later as p-helix ( hydrogen bonding). The 310 helix with 3 residues per turn and 10 atoms between the donor and the acceptor in the hydrogen bond is rarely found, except at the end of a-helices.
The a-helix content differs greatly from among proteins being present in high proportions in globular proteins like haemoglobin, myoglobin and ferritin or in fibrous proteins such as a-keratin, myosin, epidermin and fibrinogen. On the other hand it is relatively less or absent in other proteins such as chymotrypsin, superoxide dismutase and cytochrome c
Helix formers: Methionine, alanine, leucine, glutamate, and lysine ("MALEK" in the amino-acid 1-letter codes) all have especially high helix-forming propensities.
Helix breakers: Proline. At the other extreme, glycine also tends to disrupt helices because its high conformational flexibility makes it entropically expensive to adopt the relatively constrained a-helical structure.
Beta Pleated Sheet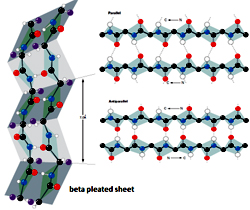 Pauling and Corey discovered the second major type of periodic secondary structure observed in proteins in 1951, the same year as the a helix, and termed ‘ß’ as it was the second structure they elucidated. The basic element is a 5 to 10 residue unit of the polypeptide, whose backbone is almost fully extended, called a ‘ß strand’, with rotation angle values of phi ~-120o and psi ~-140o. The ß strand can be regarded as a helix with only two residues per turn and a translation of 3.4 A ° per residue. A ß strand is not a stable structure and it therefore tends to interact with other ß strands that either belongs to other regions of the same polypeptide chain, distant in the primary structure (intramolecular), or are present in different polypeptide chains (intermolecular). The adjacent strands, running either with the same or opposite directions form hydrogen bonds with each other forming ß pleated sheets, which contain alternate Ca atoms lying a little above and a little below the plane of the sheet. Depending on the relative direction of the constituent ß strands (2–6 strands, on average), or else mixed ß sheets when ß strands combine with some ß strand pairs parallel and some antiparallel. In almost all known protein structures, these different b pleated sheets have a right-handed twist, with more positive values of both the phi and psi angles.
Pauling and Corey discovered the second major type of periodic secondary structure observed in proteins in 1951, the same year as the a helix, and termed ‘ß’ as it was the second structure they elucidated. The basic element is a 5 to 10 residue unit of the polypeptide, whose backbone is almost fully extended, called a ‘ß strand’, with rotation angle values of phi ~-120o and psi ~-140o. The ß strand can be regarded as a helix with only two residues per turn and a translation of 3.4 A ° per residue. A ß strand is not a stable structure and it therefore tends to interact with other ß strands that either belongs to other regions of the same polypeptide chain, distant in the primary structure (intramolecular), or are present in different polypeptide chains (intermolecular). The adjacent strands, running either with the same or opposite directions form hydrogen bonds with each other forming ß pleated sheets, which contain alternate Ca atoms lying a little above and a little below the plane of the sheet. Depending on the relative direction of the constituent ß strands (2–6 strands, on average), or else mixed ß sheets when ß strands combine with some ß strand pairs parallel and some antiparallel. In almost all known protein structures, these different b pleated sheets have a right-handed twist, with more positive values of both the phi and psi angles.
Large aromatic residues (Tyr, Phe and Trp) and ß-branched amino acids (Thr, Val, Ile) are favored to be found in ß strands in the middle of ß sheets. Interestingly, different types of residues (such as Pro) are likely to be found in the edge strands in ß sheets, presumably to avoid the "edge-to-edge" association between proteins that might lead to aggregation and amyloid formation.
ß Turns:
A third common secondary structure found in natural proteins is the ß turn (also known as a reverse turn, hairpin turn, or ß bend). The ß turns are short segments of the polypeptide chain that allow it to change direction—that is, to turn upon itself or folds back on itself by nearly 180 degrees. Turns are composed of four amino acid residues in a compact configuration in which an interamide hydrogen bond is formed between the first and fourth residue to stabilize the structure.
Tertiary Structure
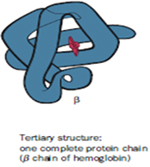 The tertiary level of structure refers to the spatial arrangement of secondary structure elements and amino acid side chain interactions of a polypeptide chain through folding and coiling to produce a three-dimensional structure. In case of some proteins, formation of tertiary structures might require the participation of molecular chaperones. Tertiary structure is generally stabilized by nonlocal interactions, most commonly the formation of a hydrophobic core, but also through salt bridges, hydrogen bonds, disulfide bonds, and even post-translational modifications. With the aid of stabilizing interactions, the secondary structures like a helix and ß strands form localized structures called Domains, consisting of 100–150 amino acid residues. The term "tertiary structure" is often used as synonymous with the term fold. Eg. Myoglobin and Lysozyme.
The tertiary level of structure refers to the spatial arrangement of secondary structure elements and amino acid side chain interactions of a polypeptide chain through folding and coiling to produce a three-dimensional structure. In case of some proteins, formation of tertiary structures might require the participation of molecular chaperones. Tertiary structure is generally stabilized by nonlocal interactions, most commonly the formation of a hydrophobic core, but also through salt bridges, hydrogen bonds, disulfide bonds, and even post-translational modifications. With the aid of stabilizing interactions, the secondary structures like a helix and ß strands form localized structures called Domains, consisting of 100–150 amino acid residues. The term "tertiary structure" is often used as synonymous with the term fold. Eg. Myoglobin and Lysozyme.
Myoglobin- The first breakthrough in understanding the three-dimensional structure of a globular protein came from x-ray diffraction studies of Myoglobin carried out by John Kendrew and his colleagues in the 1950s. Myoglobin is a relatively small, oxygen binding protein of muscle cells. It functions both to store oxygen and to facilitate oxygen diffusion in rapidly contracting muscle tissue. Myoglobin contains a single polypeptide chain of 153 amino acid residues of known sequence and a single iron protoporphyrin, or heme group. Myoglobin is particularly abundant in the muscles of diving mammals such as the whale, seal, and porpoise, whose muscles are so rich in this protein that they are brown. Storage and distributions of oxygen by muscle Myoglobin permits these animals to remain submerged for long periods of time.
Quaternary Structure
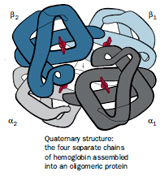 The arrangement of subunits of a protein relative to one another defines the quaternary structure of the protein. Since many proteins are made up from two or more polypeptide chains, the individual polypeptides of the active molecule are referred to as subunits. The subunits may be multiple copies of the same polypeptide chain (a homomultimer), or they may represent distinct polypeptides (a heteromultimer). In both cases the subunits fold as individual units, acquiring their own secondary and tertiary structures.
The arrangement of subunits of a protein relative to one another defines the quaternary structure of the protein. Since many proteins are made up from two or more polypeptide chains, the individual polypeptides of the active molecule are referred to as subunits. The subunits may be multiple copies of the same polypeptide chain (a homomultimer), or they may represent distinct polypeptides (a heteromultimer). In both cases the subunits fold as individual units, acquiring their own secondary and tertiary structures.
The association between subunits may be stabilized through noncovalent forces, such as hydrogen bonding, salt bridge formation, and hydrophobic interactions, and may additionally include covalent disulfide bonding between cysteines on the different subunits. On aggregation, the accessible surface area of each subunit is reduced by about 10–20%. To minimise the free energy in the aggregated form, the subunits are usually packed in a symmetrical fashion, as in crystals. Generally it is of critical importance to the proper functioning of the oligomeric proteins and except in a few cases (e.g. aspartate transcarbamylase), no protein activity is observed when the constituent subunits are separated.
Haemoglobin
Hemoglobin is a heterotetramer, composed of two a subunits and two ß subunits. Each of these four subunits contains a heme cofactor that is capable of binding a molecule of oxygen. A heme group consists of an iron (Fe) atom held in a heterocyclic ring, known as a porphyrin. The iron atom, which is the site of oxygen binding, bonds with the four nitrogens in the center of the ring. The iron is also bound strongly to the globular protein via the imidazole ring of a histidine residue below the porphyrin ring. A sixth position can reversibly bind oxygen, completing the octahedral group of six ligands. The affinity of the heme for oxygen depends on the quaternary structure of the protein and on the state of oxygen binding of the heme groups in the other three subunits.
In adult humans, the most common hemoglobin type is a tetramer (which contains 4 subunit proteins) called hemoglobin A, consisting of two a and two ß subunits non-covalently bound, each made of 141 and 146 amino acid residues, respectively. This is denoted as a2ß2. The subunits are structurally similar and about the same size. Each subunit has a molecular weight of about 17,000 daltons, for a total molecular weight of the tetramer of about 68,000 daltons. Hemoglobin A is the most intensively studied of the hemoglobin molecules.
Hemoglobin molecules almost always have all four heme sites bound to oxygen (the oxy form) or all four heme sites free of oxygen (the deoxy form); intermediate forms with one, two, or three oxygen molecules bound are almost never observed. This is due to a phenomenon called Cooperativity.
 Oxyhemoglobin (R state) is formed during respiration when oxygen binds to the heme component of the protein hemoglobin in red blood cells. This process occurs in the pulmonary capillaries adjacent to the alveoli of the lungs. The oxygen then travels through the blood stream to be dropped off at cells where it is utilized in aerobic glycolysis and in the production of ATP by the process of oxidative phosphorylation. It doesn't however help to counteract a decrease in blood pH. Ventilation, or breathing, may reverse this condition by removal of carbon dioxide, thus causing a shift up in pH.
Oxyhemoglobin (R state) is formed during respiration when oxygen binds to the heme component of the protein hemoglobin in red blood cells. This process occurs in the pulmonary capillaries adjacent to the alveoli of the lungs. The oxygen then travels through the blood stream to be dropped off at cells where it is utilized in aerobic glycolysis and in the production of ATP by the process of oxidative phosphorylation. It doesn't however help to counteract a decrease in blood pH. Ventilation, or breathing, may reverse this condition by removal of carbon dioxide, thus causing a shift up in pH.
Deoxyhemoglobin (T state) is the form of hemoglobin without the bound oxygen. The absorption spectra of oxyhemoglobin and deoxyhemoglobin differ. The oxyhemoglobin has significantly lower absorption of the 660 nm wavelength than deoxyhemoglobin, while at 940 nm its absorption is slightly higher. This difference is used for measurement of the amount of oxygen in patient's blood by an instrument called pulse oximeter.
 In the tetrameric form of normal adult hemoglobin, the binding of oxygen is thus a cooperative process. The binding affinity of hemoglobin for oxygen is increased by the oxygen saturation of the molecule, with the first oxygen bound influencing the shape of the binding sites for the next oxygen, in a way favorable for binding. This positive cooperative binding is achieved through steric conformational changes of the hemoglobin protein complex as discussed above, i.e. when one subunit protein in hemoglobin becomes oxygenated; this induces a conformational or structural change in the whole complex, causing the other subunits to gain an increased affinity for oxygen. As a consequence, the oxygen binding curve of hemoglobin is sigmoidal, or S-shaped, as opposed to the normal hyperbolic curve associated with noncooperative binding.
In the tetrameric form of normal adult hemoglobin, the binding of oxygen is thus a cooperative process. The binding affinity of hemoglobin for oxygen is increased by the oxygen saturation of the molecule, with the first oxygen bound influencing the shape of the binding sites for the next oxygen, in a way favorable for binding. This positive cooperative binding is achieved through steric conformational changes of the hemoglobin protein complex as discussed above, i.e. when one subunit protein in hemoglobin becomes oxygenated; this induces a conformational or structural change in the whole complex, causing the other subunits to gain an increased affinity for oxygen. As a consequence, the oxygen binding curve of hemoglobin is sigmoidal, or S-shaped, as opposed to the normal hyperbolic curve associated with noncooperative binding.
The sigmoidal shape of hemoglobin's oxygen-dissociation curve results from cooperative binding of oxygen to hemoglobin.
Hemoglobin's oxygen-binding capacity is decreased in the presence of carbon monoxide because both gases compete for the same binding sites on hemoglobin, carbon monoxide binding preferentially in place of oxygen. Carbon dioxide occupies a different binding site on the hemoglobin. Carbon dioxide is more readily dissolved in deoxygenated blood, facilitating its removal from the body after the oxygen has been released to tissues undergoing metabolism. This increased affinity for carbon dioxide by the venous blood is known as the Haldane effect. Through the enzyme carbonic anhydrase, carbon dioxide reacts with water to give carbonic acid, which decomposes into bicarbonate and protons:
CO2 + H2O ? H2CO3 ? HCO3- + H+
Hence blood with high carbon dioxide levels is also lower in pH (more acidic). Hemoglobin can bind protons and carbon dioxide which causes a conformational change in the protein and facilitates the release of oxygen. Protons bind at various places along the protein, and carbon dioxide binds at the alpha-amino group forming carbamate. Conversely, when the carbon dioxide levels in the blood decrease (i.e., in the lung capillaries), carbon dioxide and protons are released from hemoglobin, increasing the oxygen affinity of the protein. This control of hemoglobin's affinity for oxygen by the binding and release of carbon dioxide and acid, is known as the Bohr effect.
The binding of oxygen is affected by molecules such as carbon monoxide (CO) (for example from tobacco smoking, cars and furnaces). CO competes with oxygen at the heme binding site. Hemoglobin binding affinity for CO is 200 times greater than its affinity for oxygen, meaning that small amounts of CO dramatically reduce hemoglobin's ability to transport oxygen. When hemoglobin combines with CO, it forms a very bright red compound called carboxyhemoglobin. When inspired air contains CO levels as low as 0.02%, headache and nausea occur; if the CO concentration is increased to 0.1%, unconsciousness will follow. In heavy smokers, up to 20% of the oxygen-active sites can be blocked by CO.
In similar fashion, hemoglobin also has competitive binding affinity for cyanide (CN-), sulfur monoxide (SO), nitrogen dioxide (NO2), and sulfide (S2-), including hydrogen sulfide (H2S). All of these bind to iron in heme without changing its oxidation state, but they nevertheless inhibit oxygen-binding, causing grave toxicity.
In people acclimated to high altitudes, the concentration of 2, 3-bisphosphoglycerate (2,3-BPG) in the blood is increased, which allows these individuals to deliver a larger amount of oxygen to tissues under conditions of lower oxygen tension. This phenomenon, where molecule Y affects the binding of molecule X to a transport molecule Z, is called a heterotropic allosteric effect.
A variant hemoglobin, called fetal hemoglobin, is found in the developing fetus, and binds oxygen with greater affinity than adult hemoglobin. This means that the oxygen binding curve for fetal hemoglobin is left-shifted (i.e., a higher percentage of hemoglobin has oxygen bound to it at lower oxygen tension), in comparison to that of adult hemoglobin. As a result, fetal blood in the placenta is able to take oxygen from maternal blood.
Other oxygen-binding proteins
Myoglobin: Found in the muscle tissue of many vertebrates, including humans, it gives muscle tissue a distinct red or dark gray color. It is very similar to hemoglobin in structure and sequence, but is not a tetramer; instead, it is a monomer that lacks cooperative binding. It is used to store oxygen rather than transport it.
Hemocyanin: The second most common oxygen transporting protein found in nature, it is found in the blood of many arthropods and molluscs. Uses copper prosthetic groups instead of iron heme groups and is blue in color when oxygenated.
Hemerythrin: Some marine invertebrates and a few species of annelid use this iron containing non-heme protein to carry oxygen in their blood. It appears pink/violet when oxygenated, clear when not.
Chlorocruorin: Found in many annelids, it is very similar to erythrocruorin, but the heme group is significantly different in structure. Appears green when deoxygenated and red when oxygenated.
Vanabins: Also known as vanadium chromagens, they are found in the blood of sea squirts and are hypothesised to use the rare metal vanadium as its oxygen binding prosthetic group.
Erythrocruorin: Found in many annelids, including earthworms, it is a giant free-floating blood protein containing many dozens—possibly hundreds—of iron- and heme-bearing protein subunits bound together into a single protein complex with a molecular mass greater than 3.5 million daltons.
Pinnaglobin: Only seen in the mollusk Pinna squamosa. Brown manganese-based porphyrin protein.
Leghemoglobin: In leguminous plants, such as alfalfa or soybeans, the nitrogen fixing bacteria in the roots are protected from oxygen by this iron heme containing, oxygen binding protein.
Sickle cell anemia: The gene defect is a known mutation of a single nucleotide (A to T) of the ß-globin gene, which results in glutamic acid to be substituted by valine at position 6. Haemoglobin S with this mutation are referred to as HbS, as opposed to the normal adult HbA. The genetic disorder is due to the mutation of a single nucleotide, from a GAG to GUG codon mutation. This is normally a benign mutation, causing no apparent effects on the secondary, tertiary, or quaternary structure of haemoglobin (IISc, 2007). What it does allow for, under conditions of low oxygen concentration, is the polymerization of the HbS itself. The deoxy form of haemoglobin exposes a hydrophobic patch on the protein between the E and F helices. The hydrophobic residues of the valine at position 6 of the beta chain in haemoglobin are able to bind to the hydrophobic patch, causing haemoglobin S molecules to aggregate and form fibrous precipitates.
This process damages the red blood cell membrane, and can cause the cells to become stuck in blood vessels.
References:
The primary level of structure refers to the linear sequence of amino acids along a polypeptide chain and the location of covalent bonds, namely disulfide bonds, between chains or within a chain. Proteins can be identified explicitly based on the primary structure. In addition the chemical and biological characteristics of a protein are determined by the primary structure and it also specifies the higher levels of protein structure
Formation of the Polypeptide Chain
Amino acids are joined end to- end during protein synthesis through covalent bonds. The a-carboxyl group of the first amino acid is linked to the a-amino group of the next amino acid to generate a peptide bond and eliminate a water molecule. In this process the amino group of the first amino acid of a polypeptide chain and the carboxyl group of the last amino acid remain intact.
Therefore a polypeptide chain has two different ends, names N-terminus and C-terminus, respectively. The process of polymerization of Amino acids occurs from N-terminus to the C-terminus and is also denoted with numbers in the same direction. For example Lys48 represents a Lysine at position 18 from the amino terminus and the amino acid sequence of a polypeptide is generally written from left to right.
A polypeptide chain can be divided into two parts- the ‘main chain’ or ‘backbone’ and a variable part. The main chain comprises of a regularly repeating part forming the backbone from which the variable part arises. A variable part consists of different side chains. An amino acid unit when incorporated into a polypeptide chain is called an amino acid ‘residue’ and differs from the corresponding amino acid by lacking a water molecule. Specific names of amino acid residues are derived from the corresponding free amino acid, e.g. arginyl residue (arginine), lysyl residue (lysine) and so on.
The peptide bond –CO–NH– between two successive residues is a relatively rigid planar structure because of its partial double-bond character due to resonance. As a consequence, rotation of this bond is restricted. However, there is rotational freedom about the single bonds that link each Ca atom to the N and C atoms of peptide bonds. The angle of rotation between the N–Ca bonds is denoted as phi (f) and the angle around the Ca –C bond from the Ca –atom is denoted as psi (?). The planar peptide bond can theoretically exist in two different configurations, the trans and cis forms. The trans form is energetically favoured and is thus more stable over the cis form.
Determination of the Primary Structure
Insulin, a protein hormone of 51 residues was the first protein whose complete amino acid sequence was determined in 1953 by Frederick Sanger. The first step in determining the sequence of an amino acid is to identify the N-terminal and C-terminal residues. Chemical compounds like 1-fluoro-2, 4-dinitrobenzene or dansyl chloride or cyanate are used to label and thereby identify the N terminal residue. Likewise, the C-terminal residue can be identified by converting the a-carboxyl groups of peptide bonds into hydrazides by hydrazinolysis. Enzymatic methods causing sequential release of single amino acids from the ends of chains using specific exopeptidases, namely aminopeptidase and carboxypeptidase can also be employed.
Edman degradation is one of the most efficient procedures for determining the amino acid sequence of a protein, in which amino acids are removed sequentially, one at a time, from the N-terminus without affecting the other regions of the protein. The terminal a-amino group is first reacted with phenylisothiocyanate in alkaline conditions to yield the phenylthiocarbamoyl derivative. The derivative is then released from the rest of the chain, by acid treatment, in the form of a cyclic compound that rearranges in aqueous solution to the phenylthiohydantoin. Chromatographic techniques can be used to identify the phenylthiohydantoin derivative. The procedure can be repeated as many times as necessary to determine the complete amino acid sequence of the protein. Such recurrent analysis is usually performed in an automatic amino acid sequencer.
In case of larger proteins, the protein is initially fragmented, followed by sequencing by the Edman method. In order to execute such fragmentation in specific manner chemicals like Cyanogen bromide, hydroxylamine and N-chlorosuccinimide can be used. Alternatively enzymes like trypsin, pepsin, elastase and thermolysin can be employed. Additionally, techniques like Mass spectrometry can also be used to determining the primary structure of proteins. The technique relies on making the protein volatile by chemical treatment of side-chains and then fragmented nonspecifically with an electron beam. The protein fragments are separated according to their mass-to-charge ratio and identified. Ionisation of proteins with a high-energy beam of atoms or ions can be employed to prevent chemical modification of the protein.
Covalent Modifications of Proteins
Proteins undergo several covalent modifications during or after their biosynthesis. Proteolytic cleavage of the polypeptide chain is one such instance which involves removal of one or several residues from the polypeptide. Such modifications which attain a large biological significance are very helpful in removal of signal peptides and converting inactive form of proteins like zymogens, prohormones, proproteins and preproproteins into active forms.
Protein splicing is another mechanism where excision of internal segments (inteins) of a polypeptide occurs.
So far, about 200 modifications have been detected which can be either reversible or irreversible. The major modifications of the proteins include the addition of lipds, carbohydrates, phosphates and hydroxyl groups etc.
Addition of lipid polar groups like myristoyl groups occurs at the N- terminus and glycosyl-phosphatidylinositol and farnesyl groups at their C- terminus of proteins. Glycosylation is of two types- N-linked glycosylation and O- linked glycosylation.
The N-type occurs on the nitrogen atom of asparaginyl side-chains and O-type, on the oxygen atoms of hydroxyls, particularly those of seryl and threonyl residues.
Another key modification that specifically occurs at the hydroxyl groups is Phosphorylation. It mainly targets the hydroxyl groups of seryl, threonyl and tyrosyl residues in a wide array of proteins belonging to all biological systems and also at aspartyl and histidyl residues in the particular case of bacterial proteins. Hydroxylation is mainly targeted at prolyl and lysyl residues and is highly essential for the maturation and secretion of proteins such as collagens. Other modifications include methylation, acetylation, sulfation, Carboxylation, adenosine diphophste (ADP)-ribosylation, nucleotidylation and amidation.
Addition of non-peptidic prosthetic groups, either covalently or noncovalently like haems, porphyrins, nucleotides and metal ions is essential for some proteins like heteroproteins.
Secondary Structure
Secondary structures are the structures, which are regularly repeating local structures stabilized by hydrogen bonds. The most common examples are the alpha helix and sheet. Because secondary structures are local, many regions of different secondary structure can be present in the same protein molecule.
Prediction of Protein Secondary Structure
Hydrogen bonding interactions between the electronegative carbonyl oxygen atoms and the electropositive amide hydrogen atoms in the backbone chain of the molecule determine the secondary structures that can be adopted by polypeptides in proteins. Although several secondary structures with reasonable hydrogen bonding networks are possible theoretically, only a few possibilities are observed in polypeptides composed of L-amino acids (proteins) due to the limitation on the configuration of the backbone of each amino acid residue.
The Ramachandran Angles
Each amino acid residue had two bonds that can rotate freely generating angles that define the conformation of that residue in a protein and are called the Ramachandran angles, ? (psi) and f (phi).
The ? (psi) Angle
The bond from the a-carbon to the carbonyl group (at the C-terminus) of the amino acid residue can rotate and turn the whole plane of the amide group, which includes the carbonyl carbon, in a 360-degree range. This angle is measured by looking along that bond with the carbon of the carbonyl group in the rear and the a-carbon to the front.
The apparent angle measured between the two bonds to nitrogen can be seen as coming out of the axis of the CaC(C=O) bond. This angle is labeled ? (psi) and is measured from–180° to +180° with the positive direction being when the rear group is turned in the clockwise direction so that the rear nitrogen bond is clockwise of the front nitrogen bond (or when the front group is turned in counterclockwise direction so that the rear nitrogen bond is clockwise of the front).
The f (phi) Angle
The bond from the nitrogen (at the N-terminus) to the a-carbon of the amino acid residue can rotate and turn the whole plane of the other amide group, which includes the nitrogen, in a 360-degree range. This angle is measured by looking along that bond with the nitrogen atom in front and the a-carbon to the rear. The apparent angle measured between the two bonds to the carbonyl carbons that can be seen as coming out of the axis of the N-Ca bond. This angle is labeled f (phi) and is measured from –180° to +180° with the positive direction being the rear group is turned in clockwise direction so that the rear carbonyl bond is clockwise of the front carbonyl bond (or when the front group is turned in counterclockwise direction so that the rear carbonyl bond is clockwise of the front). The Ramachandran Plot
The ? and from f can be varied from –180° to 180° theoretically (that is 360° of rotation for each) but many combinations of these angles are almost never observed and some are very, very common in proteins. The Ramachandran plot is a plot of the torsional angles - phi (f)and psi (?) - of the residues (amino acids) contained in a peptide by plotting the f values on the x-axis and the ? values on the y-axis.Plotting the torsional angles in this way graphically shows which combination of angles are possible.
Steric Limits of ? and f
A space occupied by an atom cannot be occupied by another atom at the same time. Atoms are connected by Covalent bonds which cannot be broken but allow rotations. As mentioned earlier there are only two angles that rotate in a given residue, ? and f. But not all combinations are possible due to physical clashes of atoms in 3-dimensional space. These physical clashes are called steric interactions and the limit the available values for ? and f
The Allowed Regions in a Ramachandran Plot
The ß-sheet structures feature ? and f angle combinations of phi = -110 to -140 and psi = +110 to +135 that appear in the upper left corner of the plot. The a-helix regions have ? and f angle combinations that appear in the lower left quadrant with repeating values of phi ~-57o and psi ~-47o give a right-handed helical fold (the alpha-helix) and with values of phi=49 and psi = 26 gives a left handed a-helix. The right-handed a-helix is much more stable and is much more common in proteins. Collagen triple helix shows a phi= -51 and psi =153.These structures have ? and f angle combinations that avoid serious steric clashes between the atoms of the amide backbone and side chain groups. Glycine residues do not have the steric constraints of the other 19 residues. Proline residues are conformationally restricted due to the ring being part of the backbone.
Alpha Helix:
The a-helix structure was first predicted by Linus Pauling and Robert Corey in 1951 on the basis of crystallographic analyses of the structures of various small molecules. The a-helix is a clockwise turning or right-handed rod like structure. Right handed helix can be explained as turning in the direction of the fingers of the right hand when the thumb indicates the line of sight. Its inner part is formed by the coiled polypeptide main chain and the surface by the side-chains projecting outwards in a helical arrangement. A single turn of the helix contains 3.6 residues and, since a residue extends 1.5A °, the pitch of the helix is 5.4A °(1.5×3.6). It is stabilized by hydrogen bonds between the C=O group of each amino acid residue of the main chain and the N–H group of the residue located four residues away in the amino acid sequence This kind of repeated hydrogen bonding and repeating torsion angle values of phi ~-57o and psi ~-47o defines an a-helix. Orientation of all the hydrogen bonds and peptide groups is in the same direction, nearly parallel to the helix axis. Since each peptide bond possesses an individual dipole moment, the overall effect is a cumulative macrodipole for the helix with a positive charge at the amino end and a negative charge at the carboxyl end.
A left-handed a-helix is also possible sterically, but the side-chains are too close to the main chain and, therefore, this conformation is unstable and rarely encountered in natural polypeptides. Other types of a helix can be predict with hydrogen bonds between residues nearer together (n+3) or farther apart (n+5). The former is called the 310 helix ( hydrogen bonding) and the later as p-helix ( hydrogen bonding). The 310 helix with 3 residues per turn and 10 atoms between the donor and the acceptor in the hydrogen bond is rarely found, except at the end of a-helices.
The a-helix content differs greatly from among proteins being present in high proportions in globular proteins like haemoglobin, myoglobin and ferritin or in fibrous proteins such as a-keratin, myosin, epidermin and fibrinogen. On the other hand it is relatively less or absent in other proteins such as chymotrypsin, superoxide dismutase and cytochrome c
Helix formers: Methionine, alanine, leucine, glutamate, and lysine ("MALEK" in the amino-acid 1-letter codes) all have especially high helix-forming propensities.
Helix breakers: Proline. At the other extreme, glycine also tends to disrupt helices because its high conformational flexibility makes it entropically expensive to adopt the relatively constrained a-helical structure.
Beta Pleated Sheet
Pauling and Corey discovered the second major type of periodic secondary structure observed in proteins in 1951, the same year as the a helix, and termed ‘ß’ as it was the second structure they elucidated. The basic element is a 5 to 10 residue unit of the polypeptide, whose backbone is almost fully extended, called a ‘ß strand’, with rotation angle values of phi ~-120o and psi ~-140o. The ß strand can be regarded as a helix with only two residues per turn and a translation of 3.4 A ° per residue. A ß strand is not a stable structure and it therefore tends to interact with other ß strands that either belongs to other regions of the same polypeptide chain, distant in the primary structure (intramolecular), or are present in different polypeptide chains (intermolecular). The adjacent strands, running either with the same or opposite directions form hydrogen bonds with each other forming ß pleated sheets, which contain alternate Ca atoms lying a little above and a little below the plane of the sheet. Depending on the relative direction of the constituent ß strands (2–6 strands, on average), or else mixed ß sheets when ß strands combine with some ß strand pairs parallel and some antiparallel. In almost all known protein structures, these different b pleated sheets have a right-handed twist, with more positive values of both the phi and psi angles.Large aromatic residues (Tyr, Phe and Trp) and ß-branched amino acids (Thr, Val, Ile) are favored to be found in ß strands in the middle of ß sheets. Interestingly, different types of residues (such as Pro) are likely to be found in the edge strands in ß sheets, presumably to avoid the "edge-to-edge" association between proteins that might lead to aggregation and amyloid formation.
ß Turns:
A third common secondary structure found in natural proteins is the ß turn (also known as a reverse turn, hairpin turn, or ß bend). The ß turns are short segments of the polypeptide chain that allow it to change direction—that is, to turn upon itself or folds back on itself by nearly 180 degrees. Turns are composed of four amino acid residues in a compact configuration in which an interamide hydrogen bond is formed between the first and fourth residue to stabilize the structure.
Tertiary Structure
The tertiary level of structure refers to the spatial arrangement of secondary structure elements and amino acid side chain interactions of a polypeptide chain through folding and coiling to produce a three-dimensional structure. In case of some proteins, formation of tertiary structures might require the participation of molecular chaperones. Tertiary structure is generally stabilized by nonlocal interactions, most commonly the formation of a hydrophobic core, but also through salt bridges, hydrogen bonds, disulfide bonds, and even post-translational modifications. With the aid of stabilizing interactions, the secondary structures like a helix and ß strands form localized structures called Domains, consisting of 100–150 amino acid residues. The term "tertiary structure" is often used as synonymous with the term fold. Eg. Myoglobin and Lysozyme. Myoglobin- The first breakthrough in understanding the three-dimensional structure of a globular protein came from x-ray diffraction studies of Myoglobin carried out by John Kendrew and his colleagues in the 1950s. Myoglobin is a relatively small, oxygen binding protein of muscle cells. It functions both to store oxygen and to facilitate oxygen diffusion in rapidly contracting muscle tissue. Myoglobin contains a single polypeptide chain of 153 amino acid residues of known sequence and a single iron protoporphyrin, or heme group. Myoglobin is particularly abundant in the muscles of diving mammals such as the whale, seal, and porpoise, whose muscles are so rich in this protein that they are brown. Storage and distributions of oxygen by muscle Myoglobin permits these animals to remain submerged for long periods of time.
Quaternary Structure
The arrangement of subunits of a protein relative to one another defines the quaternary structure of the protein. Since many proteins are made up from two or more polypeptide chains, the individual polypeptides of the active molecule are referred to as subunits. The subunits may be multiple copies of the same polypeptide chain (a homomultimer), or they may represent distinct polypeptides (a heteromultimer). In both cases the subunits fold as individual units, acquiring their own secondary and tertiary structures.The association between subunits may be stabilized through noncovalent forces, such as hydrogen bonding, salt bridge formation, and hydrophobic interactions, and may additionally include covalent disulfide bonding between cysteines on the different subunits. On aggregation, the accessible surface area of each subunit is reduced by about 10–20%. To minimise the free energy in the aggregated form, the subunits are usually packed in a symmetrical fashion, as in crystals. Generally it is of critical importance to the proper functioning of the oligomeric proteins and except in a few cases (e.g. aspartate transcarbamylase), no protein activity is observed when the constituent subunits are separated.
Haemoglobin
Hemoglobin is a heterotetramer, composed of two a subunits and two ß subunits. Each of these four subunits contains a heme cofactor that is capable of binding a molecule of oxygen. A heme group consists of an iron (Fe) atom held in a heterocyclic ring, known as a porphyrin. The iron atom, which is the site of oxygen binding, bonds with the four nitrogens in the center of the ring. The iron is also bound strongly to the globular protein via the imidazole ring of a histidine residue below the porphyrin ring. A sixth position can reversibly bind oxygen, completing the octahedral group of six ligands. The affinity of the heme for oxygen depends on the quaternary structure of the protein and on the state of oxygen binding of the heme groups in the other three subunits.
In adult humans, the most common hemoglobin type is a tetramer (which contains 4 subunit proteins) called hemoglobin A, consisting of two a and two ß subunits non-covalently bound, each made of 141 and 146 amino acid residues, respectively. This is denoted as a2ß2. The subunits are structurally similar and about the same size. Each subunit has a molecular weight of about 17,000 daltons, for a total molecular weight of the tetramer of about 68,000 daltons. Hemoglobin A is the most intensively studied of the hemoglobin molecules.
Hemoglobin molecules almost always have all four heme sites bound to oxygen (the oxy form) or all four heme sites free of oxygen (the deoxy form); intermediate forms with one, two, or three oxygen molecules bound are almost never observed. This is due to a phenomenon called Cooperativity.
Oxyhemoglobin (R state) is formed during respiration when oxygen binds to the heme component of the protein hemoglobin in red blood cells. This process occurs in the pulmonary capillaries adjacent to the alveoli of the lungs. The oxygen then travels through the blood stream to be dropped off at cells where it is utilized in aerobic glycolysis and in the production of ATP by the process of oxidative phosphorylation. It doesn't however help to counteract a decrease in blood pH. Ventilation, or breathing, may reverse this condition by removal of carbon dioxide, thus causing a shift up in pH. Deoxyhemoglobin (T state) is the form of hemoglobin without the bound oxygen. The absorption spectra of oxyhemoglobin and deoxyhemoglobin differ. The oxyhemoglobin has significantly lower absorption of the 660 nm wavelength than deoxyhemoglobin, while at 940 nm its absorption is slightly higher. This difference is used for measurement of the amount of oxygen in patient's blood by an instrument called pulse oximeter.
In the tetrameric form of normal adult hemoglobin, the binding of oxygen is thus a cooperative process. The binding affinity of hemoglobin for oxygen is increased by the oxygen saturation of the molecule, with the first oxygen bound influencing the shape of the binding sites for the next oxygen, in a way favorable for binding. This positive cooperative binding is achieved through steric conformational changes of the hemoglobin protein complex as discussed above, i.e. when one subunit protein in hemoglobin becomes oxygenated; this induces a conformational or structural change in the whole complex, causing the other subunits to gain an increased affinity for oxygen. As a consequence, the oxygen binding curve of hemoglobin is sigmoidal, or S-shaped, as opposed to the normal hyperbolic curve associated with noncooperative binding.The sigmoidal shape of hemoglobin's oxygen-dissociation curve results from cooperative binding of oxygen to hemoglobin.
Hemoglobin's oxygen-binding capacity is decreased in the presence of carbon monoxide because both gases compete for the same binding sites on hemoglobin, carbon monoxide binding preferentially in place of oxygen. Carbon dioxide occupies a different binding site on the hemoglobin. Carbon dioxide is more readily dissolved in deoxygenated blood, facilitating its removal from the body after the oxygen has been released to tissues undergoing metabolism. This increased affinity for carbon dioxide by the venous blood is known as the Haldane effect. Through the enzyme carbonic anhydrase, carbon dioxide reacts with water to give carbonic acid, which decomposes into bicarbonate and protons:
CO2 + H2O ? H2CO3 ? HCO3- + H+
Hence blood with high carbon dioxide levels is also lower in pH (more acidic). Hemoglobin can bind protons and carbon dioxide which causes a conformational change in the protein and facilitates the release of oxygen. Protons bind at various places along the protein, and carbon dioxide binds at the alpha-amino group forming carbamate. Conversely, when the carbon dioxide levels in the blood decrease (i.e., in the lung capillaries), carbon dioxide and protons are released from hemoglobin, increasing the oxygen affinity of the protein. This control of hemoglobin's affinity for oxygen by the binding and release of carbon dioxide and acid, is known as the Bohr effect.
The binding of oxygen is affected by molecules such as carbon monoxide (CO) (for example from tobacco smoking, cars and furnaces). CO competes with oxygen at the heme binding site. Hemoglobin binding affinity for CO is 200 times greater than its affinity for oxygen, meaning that small amounts of CO dramatically reduce hemoglobin's ability to transport oxygen. When hemoglobin combines with CO, it forms a very bright red compound called carboxyhemoglobin. When inspired air contains CO levels as low as 0.02%, headache and nausea occur; if the CO concentration is increased to 0.1%, unconsciousness will follow. In heavy smokers, up to 20% of the oxygen-active sites can be blocked by CO.
In similar fashion, hemoglobin also has competitive binding affinity for cyanide (CN-), sulfur monoxide (SO), nitrogen dioxide (NO2), and sulfide (S2-), including hydrogen sulfide (H2S). All of these bind to iron in heme without changing its oxidation state, but they nevertheless inhibit oxygen-binding, causing grave toxicity.
In people acclimated to high altitudes, the concentration of 2, 3-bisphosphoglycerate (2,3-BPG) in the blood is increased, which allows these individuals to deliver a larger amount of oxygen to tissues under conditions of lower oxygen tension. This phenomenon, where molecule Y affects the binding of molecule X to a transport molecule Z, is called a heterotropic allosteric effect.
A variant hemoglobin, called fetal hemoglobin, is found in the developing fetus, and binds oxygen with greater affinity than adult hemoglobin. This means that the oxygen binding curve for fetal hemoglobin is left-shifted (i.e., a higher percentage of hemoglobin has oxygen bound to it at lower oxygen tension), in comparison to that of adult hemoglobin. As a result, fetal blood in the placenta is able to take oxygen from maternal blood.
Other oxygen-binding proteins
Myoglobin: Found in the muscle tissue of many vertebrates, including humans, it gives muscle tissue a distinct red or dark gray color. It is very similar to hemoglobin in structure and sequence, but is not a tetramer; instead, it is a monomer that lacks cooperative binding. It is used to store oxygen rather than transport it.
Hemocyanin: The second most common oxygen transporting protein found in nature, it is found in the blood of many arthropods and molluscs. Uses copper prosthetic groups instead of iron heme groups and is blue in color when oxygenated.
Hemerythrin: Some marine invertebrates and a few species of annelid use this iron containing non-heme protein to carry oxygen in their blood. It appears pink/violet when oxygenated, clear when not.
Chlorocruorin: Found in many annelids, it is very similar to erythrocruorin, but the heme group is significantly different in structure. Appears green when deoxygenated and red when oxygenated.
Vanabins: Also known as vanadium chromagens, they are found in the blood of sea squirts and are hypothesised to use the rare metal vanadium as its oxygen binding prosthetic group.
Erythrocruorin: Found in many annelids, including earthworms, it is a giant free-floating blood protein containing many dozens—possibly hundreds—of iron- and heme-bearing protein subunits bound together into a single protein complex with a molecular mass greater than 3.5 million daltons.
Pinnaglobin: Only seen in the mollusk Pinna squamosa. Brown manganese-based porphyrin protein.
Leghemoglobin: In leguminous plants, such as alfalfa or soybeans, the nitrogen fixing bacteria in the roots are protected from oxygen by this iron heme containing, oxygen binding protein.
Sickle cell anemia: The gene defect is a known mutation of a single nucleotide (A to T) of the ß-globin gene, which results in glutamic acid to be substituted by valine at position 6. Haemoglobin S with this mutation are referred to as HbS, as opposed to the normal adult HbA. The genetic disorder is due to the mutation of a single nucleotide, from a GAG to GUG codon mutation. This is normally a benign mutation, causing no apparent effects on the secondary, tertiary, or quaternary structure of haemoglobin (IISc, 2007). What it does allow for, under conditions of low oxygen concentration, is the polymerization of the HbS itself. The deoxy form of haemoglobin exposes a hydrophobic patch on the protein between the E and F helices. The hydrophobic residues of the valine at position 6 of the beta chain in haemoglobin are able to bind to the hydrophobic patch, causing haemoglobin S molecules to aggregate and form fibrous precipitates.
This process damages the red blood cell membrane, and can cause the cells to become stuck in blood vessels.
References:
- Lehninger Principles of Biochemistry by Albert L. Lehninger
- Fundamentals of Biochemistry by Donald Voet, Judith G. Voet
- Biochemistry by Lubert Stryer
Published date : 16 May 2014 02:16PM




More Articles


