

Sakshi Education

Enzymes are the functional units of cell metabolism. They are biological catalysts which accelerate the rate of chemical reaction while keeping themselves unchanged throughout the reaction. They do not initiate the reaction; they only form short lived complexes with the reactants. Acting in organised sequences, they catalyse the hundreds of step wise reactions by which nutrient molecules and degraded, chemical energy is conserved and transformed, and cell macromolecules are made from simple precursors. In some human diseases, especially, heritable genetic disorders, there may be a deficiency or even a total absence of one or more enzymes in the tissues. In other abnormal conditions the excessive activity of a specific enzyme can sometimes be controlled by a drug designed to inhibit its catalytic activity.
History of Enzymes
Isomerases
Vitamins used as Coenzymes.
Mechanism of Enzyme Action: Enzymes are true catalysts. They enhance the rate of specific chemical reactions. They cannot change the equilibrium point of the reactions they promote, nor are they used up or permanently changed by these reactions. They display
Initially, the substrate (or substrates, in case of enzyme-catalysed reactions involving more than one substrate) is bound at the enzyme active site, forming an enzyme–substrate (ES) complex . One of the functions of the enzyme is to orientate itself relative to the substrate. This was suggested by Fischer as a “Lock and Key” hypothesis, the enzyme being the lock and the substrate the key. The binding of substrate to the active site is via noncovalent interactions: electrostatic forces, hydrogen bonds, hydrophobic interactions, and Vanderwaals inter-atomic forces. These binding interactions lead to a precise fit between an enzyme active site and its designated substrate, which accounts for the high selectivity of enzyme catalysis. According to “Induced Fit” hypothesis proposed by Koshland, the binding of the substrate causes alteration in the geometry of the enzyme resulting in the correct orientation of the appropriate groups in the substrate and the enzyme. The process of conversion of enzyme-bound substrate to enzyme-bound product involves one or more transition states. This catalysis involves a small number of amino acid side-chains at the “Active site” of the enzyme. An active site is the cleft or depression present in the enzyme where catalysis takes place and is lined with mainly hydrophobic amino acids into which the substrate fits. These amino acids impart the specifically to the enzyme and are also involved in catalysis. These amino acids which participate in the reaction are mostly acidic or basic groups or in some cases are nucleophilic groups that form a covalent bond with the substrate at an intermediate stage in the reaction. They mainly include amino acids like His, Lys, Cys, Ser, .Asp and Glu. Many enzyme reactions are a consequence of either nucleophilic or electrophilic attack on the substrate, the former being the most common. Finally, the enzyme–product (EP) complex dissociates to release the product P, and regenerate the active enzyme, which is free to catalyse another turnover of the same reaction. Some enzyme-catalysed reactions also require the presence of a small cofactor.
In order to understand the mechanism of enzyme action we need to determine-
Control at the Post-translational Level by Proteolytic Cleavage: Enzymes like proteases are synthesized as inactive precursors that require posttranslational modifications so as to form the active enzyme. Cleavage of a propeptide sequence at the amino-terminal end of the protein by a separate protease generates the active form of the enzyme. A cascade of protein cleavage steps is involved in the blood clotting response in mammals.
Control at the post translational level by chemical modifications: Modifications like phosphorylation or glycosylation at specific sites produce a phosphoenzyme or glycoprotein with increased or decreased activity. For example glycogen breakdown by the enzyme glycogen phosphorylase is regulated by phosphorylation.
Control by means of allosteric sites: Activation of an enzyme can occur in response to binding of a effector molecule to an site in the enzume apart from active site. Such a site is called Allosteric site. An example is phosphofructokinase is regulated by ATP, AMP and citrate.
History of Enzymes
- The word “Enzymes” is derived from the Greek meaning “in yeast” and was first used by Kuhne in 1878.
- Emil Fischer (1894) proposed the “Lock and key” mechanism for the specificity of the enzyme to its substrate.
- In 1897 Eduard Buchner published the observation that the cell free extracts of yeast containing in living cells were able to carry out the fermentation of sugar to alcohol and carbondioxide. He proposed a species called Zymase found in yeast cells was responsible for fermentation.
- The biochemical pathway involved in fermentation was subsequently elucidated by Embden and Meyerhof- the first pathway to be discovered.
- J. Sumner (1926) crystallised the first enzyme urease from jack beans, which hydrolyses urea to CO23.
- Koshland (1958) proposed the “Induced fit” theory to account for the catalytic power and specificity shown by enzymes.
- Monod et al proposed the “Allosteric model” to explain in a quantitative way the regulation of catalysis of certain enzymes by the binding of small molecules (effectors).
- First chemical synthesis of enzyme (Ribonuclease) from amino acid precursors by Gutte & Merrified (1969). However, the chemical purity and catalytic activity were rather low.
Nomenclature and Classification of Enzymes:
The accepted nomenclature of enzymes by the International Union of Biochemistry shows the type of reaction catalysed and described the substrate (s) acted on and any other information. Each enzyme is assigned a four digit classification number and a systematic name.
Systematic name attempts to describe in unambiguous terms the reaction that the enzyme catalyses. Systematic names consist of two parts. The first contains the name of the substrate or, in the case of a bimolecular reaction, of the two substrates separated by a colon. The second part, ending in -ase, indicates the nature of the reaction. A number of generic words indicating a type of reaction may be used in either common or systematic names: oxidoreductase, oxygenase, transferase (with the prefix indicating the nature of the group transferred), hydrolase, lyase, racemase, epimerase, isomerase, mutase, ligase.Where additional information is needed to make the reaction clear, a phrase indicating the reaction or a product should be added in parentheses after the second part of the name, e.g.(ADP-forming), (dimerizing),(CoA-acylating).
For example-ATP: glucose phosphotransferase is given a classification Number: 2.7.1.1.
This indicates that the enzymes catalyses the reaction.
[ATP + D-glucose ADP + D-glucose-6-phosphate]The first digit (2) of number stands for the Name of the class – Transferase.
The second digit (7) for the subclass – Phosphotransferase.
Transferring phosphorus-containing groups
2.7.1. – Phosphotransferases with an alcohol group as acceptor
2.7.2. – Phosphotransferases with a carboxyl group as acceptor
2.7.3. – Phosphotransferases with a nitrogenous group as acceptor
2.7.4. – Phosphotransferases with a phosphate group as acceptor
2.7.5. – Phosphotransferases with regeneration of donors, catalyzing intramolecular transfers
2.7.6. – Diphosphotransferases
2.7.7. – Nucleotidyltransferases
2.7.8. – Transferases for other substituted phosphate groups
2.7.9. – Phosphotransferases with paired acceptors
The third digit (1) for the sub-subclasses (phosphotransferase with a hydroxyl group as acceptor) and the fourth digit (1) for D-glucose as the phosphate group acceptor. When the systematic name of an enzyme is long or cumbersome, a trivial name may be used. In this case, it is ‘Hexokinase’. Most enzymes catalyse the transfer of electrons, atoms or functional groups. They are classified, given code numbers and assigned names according to the type of transfer reaction, the group donor, and the group acceptor.
There are six major types of enzymes based on their catalyze.
The accepted nomenclature of enzymes by the International Union of Biochemistry shows the type of reaction catalysed and described the substrate (s) acted on and any other information. Each enzyme is assigned a four digit classification number and a systematic name.
Systematic name attempts to describe in unambiguous terms the reaction that the enzyme catalyses. Systematic names consist of two parts. The first contains the name of the substrate or, in the case of a bimolecular reaction, of the two substrates separated by a colon. The second part, ending in -ase, indicates the nature of the reaction. A number of generic words indicating a type of reaction may be used in either common or systematic names: oxidoreductase, oxygenase, transferase (with the prefix indicating the nature of the group transferred), hydrolase, lyase, racemase, epimerase, isomerase, mutase, ligase.Where additional information is needed to make the reaction clear, a phrase indicating the reaction or a product should be added in parentheses after the second part of the name, e.g.(ADP-forming), (dimerizing),(CoA-acylating).
For example-ATP: glucose phosphotransferase is given a classification Number: 2.7.1.1.
This indicates that the enzymes catalyses the reaction.
[ATP + D-glucose ADP + D-glucose-6-phosphate]The first digit (2) of number stands for the Name of the class – Transferase.
The second digit (7) for the subclass – Phosphotransferase.
Transferring phosphorus-containing groups
2.7.1. – Phosphotransferases with an alcohol group as acceptor
2.7.2. – Phosphotransferases with a carboxyl group as acceptor
2.7.3. – Phosphotransferases with a nitrogenous group as acceptor
2.7.4. – Phosphotransferases with a phosphate group as acceptor
2.7.5. – Phosphotransferases with regeneration of donors, catalyzing intramolecular transfers
2.7.6. – Diphosphotransferases
2.7.7. – Nucleotidyltransferases
2.7.8. – Transferases for other substituted phosphate groups
2.7.9. – Phosphotransferases with paired acceptors
The third digit (1) for the sub-subclasses (phosphotransferase with a hydroxyl group as acceptor) and the fourth digit (1) for D-glucose as the phosphate group acceptor. When the systematic name of an enzyme is long or cumbersome, a trivial name may be used. In this case, it is ‘Hexokinase’. Most enzymes catalyse the transfer of electrons, atoms or functional groups. They are classified, given code numbers and assigned names according to the type of transfer reaction, the group donor, and the group acceptor.
There are six major types of enzymes based on their catalyze.
- Oxido-reductase - Transfer of electrons
- Transferases - Group transfer reactions
- Hydrolases - Hydrolysis reaction (transfer of functional group to water)
- Lyases - Addition of groups in double bonds or the reverse
- Isomerases - Transfer of groups within molecules to yield isomeric forms
- Ligases - Formation of C-C,C-S,C-O,C-N, bonds by condensation reaction coupled to ATP cleavage
- Alcohol dehydrogenase (1.1.1.1) Alcohol + NAD+ Aldehyde/kexne +NADH. .
- Glyceraldehyde-3- phosphate dehydrogenase (1.2.1.1.2) D-Glyceraldehyd-3-phosphate + orthophosphate + NAD+ 3-Phospho- D- glyceroyl phosphate + NADH. 3.
- Malate dehydrogenase (1.1.1.37) L- malate + NAD+ Oxaloactate +NADH 4. Malate dehydrogenase (1.1.1.40) L- malate + NADP+ Pyruvate + CO2 + NADPH 5.
- NADH dehydrogenase (1.6.99.3) NADH + acceptor NAD+ + reduced acceptor.
- Adenylate kinase (2.7.4.3) ATP +AMP 2ADP
- Alanine aminotransferase (2.6.1.2) L-alanine + 2-oxoglutarate Pyruvate + L-Glutamate.
- Galactose-1- Phosphate Uridylyl transferase (2.7.7.10) UTP + a-D-Galactose-1-phosphate Pyrophosphate +UDP Galactose.
- Glucokinase (2.7.1.2) ATP + D-glucose ADP + D-Glucose phosphate
- Hexokinase (2.7.1.1) ATP + D-Hexose ADP + D-Hexose-6-phosphate
- Adenosine triphosphatase 3.6.1.3 ATP + H2O ADP + Orthophosphate
- a - Amylase 3.2.1.1 Endohydrolysis of 1, 4- D-glucosidic linkages in polysaccharides.
- b -amylase 3.2.1.1. Endohydrolysis of 1, 4- a -D- glucosidic linkages to remove successine maltose units.
- Cellulase 3.2-1-4. Endohydrolysis of 1, 4- b -D-Glycosidic linkage in cellulose, lichenin, etc.
- Glucose-6- phosphatase 3.1.3.9. D-Glucose- 6- phosphate + H2O D-glucose + orthophosphate.
- Aconitase 4.2.1.3. Citrate Cis-aconitate + H2O
- Enolase (4.2.1.11)2- Phospho-D-glycerate P=P + H2O
- Ribulose biphosphate carboxylase 4.1.1.39. D-Ribulose 1,5-biphosphate + CO2 2, 3- Phospho-D-glycerate
- Isocitrate lyase 4.1.3.1 Isocitrate Succinate +Glycocylate
- Ornithine decarboxylase 4.1.1.17.L-ornithine Putrescine + CO2
Isomerases
- Aldolase-1- epimerase 5.1.3.3. a -D-Glucose=ß-D-Glucose
- Glucose-6-phosphate isomerase 5.3.1.9. D-Glucose-6-phosphate D-Fructose-6-Phosphate
- Phosphoglucomutase 5.4.2.2. a -D-Glucose 1,6- biphosphate + a -D-Glucose 1- phosphate a-D-Glucose 6- phosphate + a -D-Glucose 1,6- biphosphate.
- Phospho glycerate m lactose 5.4.2.12- Phospho- D-Glycerate 3-Phospho-D-glycerate
- Protein disulphide isomerase 5.3.4.2. Rearranges the 5-5. Bonds in protiens.
- Pyruvate carboxylase 6.4.1.1 ATP +Pyruvate + CO2 +H2O ADP + Orthophosphate + oxaloacetate.
- Polydeoxyribonucleotide synthetase (DNA Ligase) 6.5.1.2.NAD+ + (deoxyribonucleotide)n + (deoxyribonucleotide)m AMP + NMN + (deoxyribonucleotide)m+n
- Argininosuccinate synthetase 6.3.4.5.ATP + L-citrulline + aspartate AMP + Pyrophosphate + L-Arginiosuccinate
- Aminoacyl + RNA synthetases 6.1.1 (1-7 9-12, 14-22) ATP + L-aminoacid + RNA AMP + Pyrophosphate + L-aminoacid + RNA
Nature of Enzymes Enzymes are mostly protein macromolecules of molecular mass 10,000–10,00,00 Da, and therefore by means of protein folding process, acquire a large, globular three-dimensional structure in aqueous solution. A part of this structure, known as the active site, is where catalysis takes place. The active site is often a cleft or cavity in the protein architecture that contains hydrophobic (‘water-hating’) regions that exclude the presence of solvent water. As a consequence of protein folding, the amino acid residues that protrude into the active site of the enzyme often arise from quite different parts of the primary sequence of the polypeptide. For example, the three catalytic residues at the active site of the serine protease a-chymotrypsin are serine195, histidine57, aspartic acid102, which in the tertiary structure of the enzyme are aligned in a precise geometrical arrangement in order to participate in catalysis. Since enzymes are mostly proteins in nature, depends upon the integrity of the three dimensional structure determines their catalytic activity. Changes in the three dimensional structure can lead to destabilization and loss of enzymatic activity and such a process is called Denaturation. Agents like heat, acid and denature enzymes and are termed as denaturants. For example if an enzyme is boiled with strong acid can cause denaturation. Similarly treatment with proteases like trypsin which cleave the polypeptide chains causes loss of catalytic activity revealing the fact that the primary backbone (amino acid sequence) is required for the activity. Additionally, denaturants like heat, extreme pH, Urea and SDS disrupt the characteristic folding of the polypeptide chain of a native enzyme causing loss of catalytic activity affecting the primary, secondary and tertiary structures of the enzymes revealing their essential role for enzymatic activity. Enzymes are larger than their substrates or functional group they act on where some enzymes consist of only polypeptides eg. Pancreatic ribonuclease and some enzymes require an additional chemical component i.e. “Cofactor” for activity. The cofactor may be inorganic such as Fe+2, Mn+2 or Zn+2 Ions or it may be a complex organic molecule- “Co-enzyme”, some enzymes require both enzymes and one or more cofactors for their activity. In some enzymes the cofactor or coenzyme is loosely and transiently bound to the protein; but in others it is tightly and permanently bound and is called a “Prosthetic group”. A complete catalytically active enzyme together with its coenzyme or cofactor is called “Holoenzyme”. Coenzymes and cofactors are heat stable where as the protein part “Apoenzyme” is denatured by heat. The coenzyme functions as transient carriers of specific functional groups. Coenzymes were discovered as a class of biologically important small molecules that had remarkable properties in curing certain dietary disorders. These molecules were christened the vitamins. Many of the important dietary vitamins are coenzymes that are used by certain types of enzyme in order to carry out particular classes of reaction. Co-factors
| Metal | Enzymes |
| Na+ | Intestinal sucrose a-D-gluco-hydrolase |
| K+ | Pyruvate kinase |
| Mg++ | Kinase |
| Fe++ or Fe+++ | Catalase, Peroxidases, Nitrogenase |
| Zn++ | Alcohol dehydrogenase, Carboxy peptidase |
| Mo | Xanthine oxidase, Amine peroxidise |
Vitamins used as Coenzymes.
| Vitamin | Chemical Name | Deficiency Disease | Associated Chemistry | |
| B1 | Thiamin | Beriberi | Decarboxylation of a-keto acids | |
| B2 | Riboflavin | Skin lesions | Oxidation/reduction chemistry | |
| Niacin | Nicotinamide | Pellagra | Oxidation/reduction chemistry | |
| B6 | Pyridoxal | Convulsions | Reactions of a-amino acids | |
| B12 | Cobalamin | Pernicious anaemia | Skeletal rearrangements | |
| C | Ascorbic acid | Scurvy | Reducing agent (collagen formation) | |
| H | Biotin | Skin lesions | Carboxylation | |
| K | Phylloquinone | Bleeding disorders | Carboxylation of glutamyl peptides | |
| Folic acid | Megaloblastic anaemia | Transfer of 1-carbon units | ||
| Pantothenic acid | Burning foot syndrome | Acyl-transfer reactions |
Mechanism of Enzyme Action: Enzymes are true catalysts. They enhance the rate of specific chemical reactions. They cannot change the equilibrium point of the reactions they promote, nor are they used up or permanently changed by these reactions. They display
- High catalytic power
- Specifically
- Regulation by a number of naturally occurring compounds
Initially, the substrate (or substrates, in case of enzyme-catalysed reactions involving more than one substrate) is bound at the enzyme active site, forming an enzyme–substrate (ES) complex . One of the functions of the enzyme is to orientate itself relative to the substrate. This was suggested by Fischer as a “Lock and Key” hypothesis, the enzyme being the lock and the substrate the key. The binding of substrate to the active site is via noncovalent interactions: electrostatic forces, hydrogen bonds, hydrophobic interactions, and Vanderwaals inter-atomic forces. These binding interactions lead to a precise fit between an enzyme active site and its designated substrate, which accounts for the high selectivity of enzyme catalysis. According to “Induced Fit” hypothesis proposed by Koshland, the binding of the substrate causes alteration in the geometry of the enzyme resulting in the correct orientation of the appropriate groups in the substrate and the enzyme. The process of conversion of enzyme-bound substrate to enzyme-bound product involves one or more transition states. This catalysis involves a small number of amino acid side-chains at the “Active site” of the enzyme. An active site is the cleft or depression present in the enzyme where catalysis takes place and is lined with mainly hydrophobic amino acids into which the substrate fits. These amino acids impart the specifically to the enzyme and are also involved in catalysis. These amino acids which participate in the reaction are mostly acidic or basic groups or in some cases are nucleophilic groups that form a covalent bond with the substrate at an intermediate stage in the reaction. They mainly include amino acids like His, Lys, Cys, Ser, .Asp and Glu. Many enzyme reactions are a consequence of either nucleophilic or electrophilic attack on the substrate, the former being the most common. Finally, the enzyme–product (EP) complex dissociates to release the product P, and regenerate the active enzyme, which is free to catalyse another turnover of the same reaction. Some enzyme-catalysed reactions also require the presence of a small cofactor.
In order to understand the mechanism of enzyme action we need to determine-
- The elementary steps of the overall reaction i.e. the sequence in which the enzyme containing complexes and substrates are converted into products,
- The rates at which these complexes are inter converted and
- The structures of these complexes.
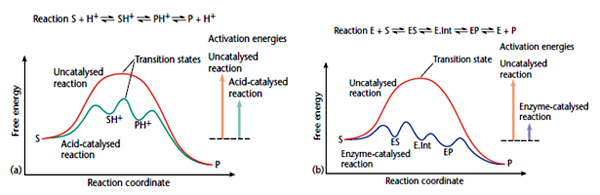
Enzymes enhance the rate of chemical reactions by lowering their Activation energy
A chemical reaction such as X -->Y can occur as X molecule at any given point have higher internal energy compared to the remaining of the population, just enough to bring them to the top of ‘Energy Hill’ to reactive form called ‘Transition State’. The amount of energy (in calories) needed to bring all the molecules in one mole of substrate to the transition state at the top of the energy barrier, at a given temperature are termed as the activation energy of reaction. When the activation energy required to reach a transition state for a large fraction of a molecule X that form a complex EX is much lower compared to the transition state of X alone, the rate of a chemical reaction will be high. The complex thus formed reacts subsequently to give rise to the product Y, releasing the free enzyme which can combine with another molecule of X and repeat the cycle. Thus, the enzyme lowers the activation energy of chemical reactions, allowing a larger fraction of the molecules to react per unit time.
 Enzymes enhance the rate of chemical reactions by transition state stabilization.
Enzymes enhance the rate of chemical reactions by transition state stabilization.
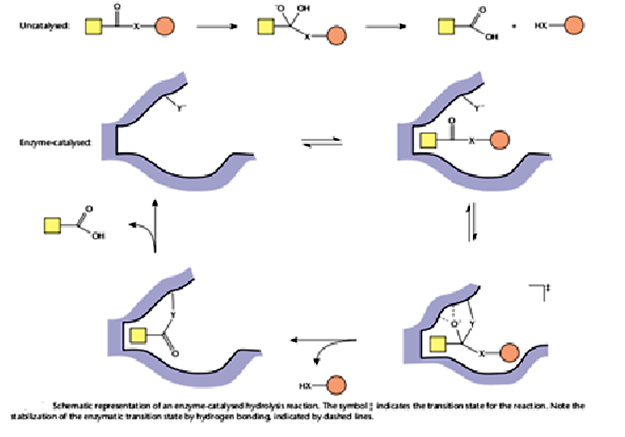
There are specific interactions that are formed between the enzyme active site and the transition state of the reaction but that are formed with neither the ES nor EP complexes. For example, a specific hydrogen bond might be formed with the transition state as a bond breaks. The high-energy transition state is stabilized in this manner and thereby reduces the activation energy, so as to accelerate the reaction. Attack of a nucleophilic active site group Y generates a transition state which is selectively stabilized by formation of hydrogen bonds with the enzyme, and by shape complementarity with the enzyme. A covalent acyl-enzyme intermediate is then formed, which is hydrolysed to regenerate free enzyme. This type of catalysis is used by the serine and cysteine proteases (e.g. a- chymotrypsin and papain, respectively), in which the group Y is either the hydroxyl side-chain of a serine residue, or a thiol side-chain of a cysteine residue.
 Catalysis by RNA:
Catalysis by RNA:
As mentioned earlier majority of the enzymes are proteins but not all. However, it has been proved that ribonucleic acid (RNA) also possess the ability to catalyse various biochemical reactions. Thomas Cech and Altman showed that the excision of certain introns in Tetrahymena is due to self splicing by RNA itself.. In addition it has been also shown that, RNA as an integral part of the cellular ribosome catalyses the assembly of protein from an RNA template, using amino acyl-tRNA molecules as substrates.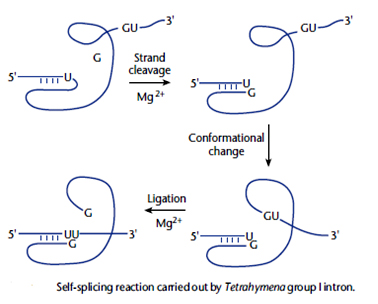
Mechanisms of Enzyme Catalysis
A chemical reaction such as X -->Y can occur as X molecule at any given point have higher internal energy compared to the remaining of the population, just enough to bring them to the top of ‘Energy Hill’ to reactive form called ‘Transition State’. The amount of energy (in calories) needed to bring all the molecules in one mole of substrate to the transition state at the top of the energy barrier, at a given temperature are termed as the activation energy of reaction. When the activation energy required to reach a transition state for a large fraction of a molecule X that form a complex EX is much lower compared to the transition state of X alone, the rate of a chemical reaction will be high. The complex thus formed reacts subsequently to give rise to the product Y, releasing the free enzyme which can combine with another molecule of X and repeat the cycle. Thus, the enzyme lowers the activation energy of chemical reactions, allowing a larger fraction of the molecules to react per unit time.
Enzymes enhance the rate of chemical reactions by transition state stabilization.There are specific interactions that are formed between the enzyme active site and the transition state of the reaction but that are formed with neither the ES nor EP complexes. For example, a specific hydrogen bond might be formed with the transition state as a bond breaks. The high-energy transition state is stabilized in this manner and thereby reduces the activation energy, so as to accelerate the reaction. Attack of a nucleophilic active site group Y generates a transition state which is selectively stabilized by formation of hydrogen bonds with the enzyme, and by shape complementarity with the enzyme. A covalent acyl-enzyme intermediate is then formed, which is hydrolysed to regenerate free enzyme. This type of catalysis is used by the serine and cysteine proteases (e.g. a- chymotrypsin and papain, respectively), in which the group Y is either the hydroxyl side-chain of a serine residue, or a thiol side-chain of a cysteine residue.
Catalysis by RNA: As mentioned earlier majority of the enzymes are proteins but not all. However, it has been proved that ribonucleic acid (RNA) also possess the ability to catalyse various biochemical reactions. Thomas Cech and Altman showed that the excision of certain introns in Tetrahymena is due to self splicing by RNA itself.. In addition it has been also shown that, RNA as an integral part of the cellular ribosome catalyses the assembly of protein from an RNA template, using amino acyl-tRNA molecules as substrates.
Mechanisms of Enzyme Catalysis
- Approximation (Proximity and orientation) of Reactants: Enzymes by bringing the substrates into close proximity to the catalytic site enhance the rate of reaction and also aid in the proper orientation so that the reaction is feasible. For instance, if two molecules X and Y in a aqueous system (like cell) react to form X-Y, then they have overcome several factors such as: (1) encounter each other through diffusion-limited collisions in the correct mutual orientations for reaction; (2) undergo changes in solvation that allow for molecular orbital interactions; (3) overcome Vanderwaals repulsive forces; and (4) undergo changes in electronic orbitals to attain the transition state configuration. In an enzyme catalysed reaction, the substrates are sequestered within the active site of the enzyme; their effective concentrations are greatly increased with respect to their concentrations in solution. A second aspect of approximation effects is that the structure of the enzyme active site is designed to bind the substrates in a specific orientation that aids in attaining the transition state, optimal for reaction.
- General Acid-base Catalysis: Many reactions catalysed by enzymes are known to be catalysed by acids /or bases since enzymes contain a number of amino acids with side chains that are capable of acting as proton donors or acceptors.In such cases, protons (from hydronium ion H3O+) and hydroxide ions (OH--) act directly as the acid and base groups in activities referred to as specific acid and specific base catalysis. Most often, however, in enzymatic reactions organic substrates, cofactors, or amino acid side chains from the enzyme fulfill this role by acting as Brønsted—Lowry acids and bases in what is referred to as general acid and general base catalysis. The fundamental feature of general acid/base catalysis is that the catalytic group participates in proton transfers that stabilize the transition state of the chemical reaction.
- Covalent Catalysis: There are numerous examples of enzyme-catalyzed reactions that go through the formation of a covalent intermediate between the enzyme and the substrate. Several families of enzymes have been demonstrated to form covalent intermediates, including serine proteases (acyl—serine intermediates), cysteine proteases (acyl—cysteine intermediates), protein kinases and phosphatase (phospho—amino acid intermediates), and pyridoxal phosphate-utilizing enzymes (pyridoxal—amino acid Schiff bases) bringing the reaction system towards the transition state, thus helping to overcome the activation energy barrier. Covalent catalysis in enzymes is facilitated mainly by nucleophilic and electrophilic catalysis, and in more specialized cases by redox catalysis.
- Conformational Distortion or Strain: Binding of the substrate may induce conformational change in the enzyme molecule which strains the structure of the active site and also distorts the bound substrate. Thus help to bring the ES complex into its transition state. Three major models have been put forth to fill these needs: (1) the induced-fit model, (2) the nonproductive binding model, and (3) the induced-strain model.
- Preorganization of the active site for transition state complementarity: An alternative to mechanisms utilizing conformational distortion is one in which the enzyme active site is, relatively speaking, conformationally rigid and preorganized to optimally fit the substrate in its transition state conformation. This is somewhat reminiscent of the lock and key model of Fischer, but here the complementarity is with the substrate transition state, rather than the ground state.
- Changes in Environment: X-ray crystallographic studies show that enzymes are capable of providing unusual environment for reactions contributing to the enhancement of rate of reaction.
Control at the Post-translational Level by Proteolytic Cleavage: Enzymes like proteases are synthesized as inactive precursors that require posttranslational modifications so as to form the active enzyme. Cleavage of a propeptide sequence at the amino-terminal end of the protein by a separate protease generates the active form of the enzyme. A cascade of protein cleavage steps is involved in the blood clotting response in mammals.
Control at the post translational level by chemical modifications: Modifications like phosphorylation or glycosylation at specific sites produce a phosphoenzyme or glycoprotein with increased or decreased activity. For example glycogen breakdown by the enzyme glycogen phosphorylase is regulated by phosphorylation.
Control by means of allosteric sites: Activation of an enzyme can occur in response to binding of a effector molecule to an site in the enzume apart from active site. Such a site is called Allosteric site. An example is phosphofructokinase is regulated by ATP, AMP and citrate.
Published date : 26 May 2014 02:29PM




More Articles


