

Sakshi Education
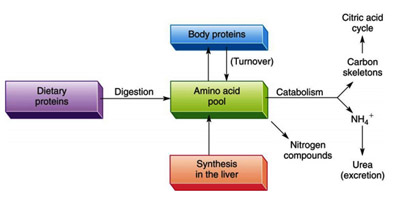
The components of living cells are constantly turning over. In the same manner, proteins also have lifetimes that range from as short as a few minutes to weeks or more. In any case, cells continuously synthesize proteins from and degrade them to aminoacids.
The process in which body proteins are continuously hydrolyzed and resynthesized is called protein turnover. The life expectancy, of body proteins is a measure of how fast they are broken down and resynthesized, expressed as a half-life. However, controlling a protein’s rate of degradation is therefore as important to the cellular and organismal economy as is controlling its rate of synthesis.
Protein degradation by non-specific mechanism
Generally “non-specific” protein degradation takes place in lysosomes – specialized organelles that operate at low pH (to denature proteins) and contain proteases for proteins, lipases for lipids, and many other hydrolases (~ 50 total). Both internal proteins (enclosed in vacuoles that fuse with lysosomes) and external proteins (obtained via endocytosis) are transported to lysosomes where proteins are degraded and the resulting amino acids either recycled for new protein synthesis or degraded for energy production or storage. In normal healthy cells, lysosomal protein degradation is nonselective. However, lysosomes in tissues that waste away or atrophy during starvation (e.g., liver and muscle, but thankfully not brain and testes) have a special degradative pathway activated only during prolonged fasting – this involves selective degradation of cytosolic proteins containing the pentapeptide sequence Lys-Phe-Glu-Arg-Gln (KFERQ), thus sparing more essential proteins from increased non-selective degradation. Such KFERQ proteins are selectively lost from tissues that atrophy in response to fasting (e.g., liver and kidney) but not from tissues that do not do so (e.g., brain and testes).
Protein degradation by specific mechanism
Although most control of metabolic activity and cell function is at the level of post-translational control or action of regulatory molecules, some control is at the actual level of the proteins themselves – by synthesis and/or degradation. This controlled or programmed protein degradation involves the Ubiquitin-Proteasome system.
This process occurs in three steps:
In the mitochondria oxaloacetate is tranaminated by glutamate by aspartate amino transferase to produce the aspartate which is then transported into the cytosol where it is required for the argininosuccinate synthetase reaction. These reactions together are called the aspartate-argininosuccinate shunt. The summary of the links between the urea cycle and the citric acid cycle is shown above diagram.
Regulation of urea cycle
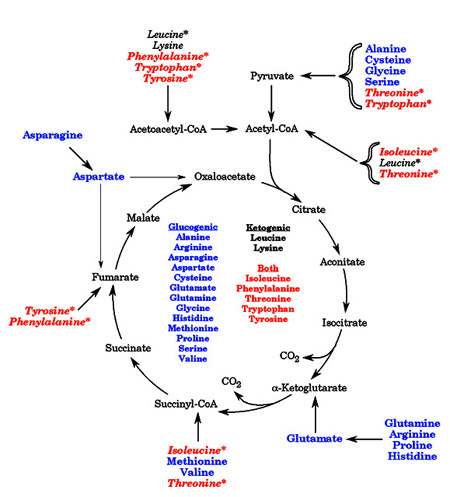 Free amino acids are not stored. Therefore, in all organisms, amino acid breakdown occurs whenever amino acid levels exceed requirements for synthetic processes Amino acid catabolism is critically important. There are two reasons for this: 1) amino acids are a potential source of energy, and especially during fasting, of glucose, and 2) incomplete metabolism of a number of amino acids results in accumulation of toxic amino acid breakdown intermediates. When considering their degradation, amino acids are generally divided into two classes. The term glucogenic refers to amino acids with a carbon skeleton that can be converted to a gluconeogenic or TCA cycle intermediate. These amino acids can be used to synthesize glucose. The term ketogenic refers to amino acids with a carbon skeleton that can only be converted to acetyl CoA, to acetoacetyl-CoA, or to acetoacetate. The ketogenic amino acids are a potential source of ketone bodies (hence the name “ketogenic”), but cannot be used to synthesize glucose. Some amino acids fall into both categories. The diagram below summarizes the conversion of the twenty standard amino acids into compounds that can be used for energy. Note that some of the amino acids are converted into more than one metabolite; these amino acids are indicated by an asterisk in the figure
Free amino acids are not stored. Therefore, in all organisms, amino acid breakdown occurs whenever amino acid levels exceed requirements for synthetic processes Amino acid catabolism is critically important. There are two reasons for this: 1) amino acids are a potential source of energy, and especially during fasting, of glucose, and 2) incomplete metabolism of a number of amino acids results in accumulation of toxic amino acid breakdown intermediates. When considering their degradation, amino acids are generally divided into two classes. The term glucogenic refers to amino acids with a carbon skeleton that can be converted to a gluconeogenic or TCA cycle intermediate. These amino acids can be used to synthesize glucose. The term ketogenic refers to amino acids with a carbon skeleton that can only be converted to acetyl CoA, to acetoacetyl-CoA, or to acetoacetate. The ketogenic amino acids are a potential source of ketone bodies (hence the name “ketogenic”), but cannot be used to synthesize glucose. Some amino acids fall into both categories. The diagram below summarizes the conversion of the twenty standard amino acids into compounds that can be used for energy. Note that some of the amino acids are converted into more than one metabolite; these amino acids are indicated by an asterisk in the figure
The amino acids are also frequently divided into families, based on the type of amino acid, or the final product formed from the amino acid.
Synthesis of amino acids
 Synthesis and/or collection of amino acids is critical for cell survival. They not only serve as the building blocks for proteins but also as starting points for the synthesis of many important cellular molecules including vitamins and nucleotides. In most cases bacteria would rather use amino acids in their environment than make them from scratch. It takes a considerable amount of energy to make the enzymes for the pathway as well as the energy required to drive some of the reactions of amino acid biosynthesis. The genes that code for amino acid synthesis enzymes and the enzymes themselves are under tight control and are only turned on when they are needed.
Synthesis and/or collection of amino acids is critical for cell survival. They not only serve as the building blocks for proteins but also as starting points for the synthesis of many important cellular molecules including vitamins and nucleotides. In most cases bacteria would rather use amino acids in their environment than make them from scratch. It takes a considerable amount of energy to make the enzymes for the pathway as well as the energy required to drive some of the reactions of amino acid biosynthesis. The genes that code for amino acid synthesis enzymes and the enzymes themselves are under tight control and are only turned on when they are needed.
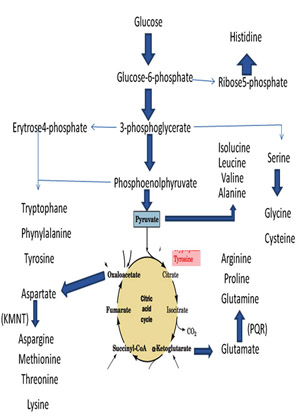
The amino acids synthesis pathways can be grouped into several logical units. These units reflect either common mechanisms or the use of common enzymes that synthesize more than one amino acid. These categories are: simple reactions, branch chain amino acids, aromatic amino acids, threonine/lysine, serine/glycine, and unique pathways. The aromatic amino acids, threonine/lysine and serine/glycine pathways have a common beginning and then diverge to form the amino acid of interest.
Notice that each pathway begins with a central metabolite or something derived from "central metabolism". Using common compounds instead of synthesizing them from scratch saves energy and conserves genes since fewer enzymes are needed to code for the pathways. Most of standard amino acids are synthesized by glycolysis and citric acid cycle is shown below.
Inborn errors of amino acid metabolism
An inability to degrade amino acids causes many genetic diseases in humans. These diseases include phenylketonuria (PKU), which results from an inability to convert phenylalanine to tyrosine. The phenylalanine is instead transaminated to phenylpyruvic acid, which is excreted in the urine, although not fast enough to prevent harm. PKU was formerly a major cause of severe mental retardation. Now, however, public health laboratories screen the urine of every newborn child in the United States for the presence of phenylpyruvate, and place children with the genetic disease on a synthetic low-phenylalanine diet to prevent neurological damage.
Heme biosynthesis
Inborn metabolic diseases that interfere with heme biosynthesis are called porphyrias. Porphyrias have a variety of symptoms. A deficiency in the enzyme responsible for the condensation of porphobilinogen to the 4-membered ring system leads to a condition called acute intermittent porphyria, which is characterized by occasional episodes of abdominal pain and psychiatric symptoms. Defects in the later enzymes of the pathway lead to an excess accumulation of the uroporphobilinogens in the tissues, where they cause a variety of symptoms, including hairy skin, skeletal abnormalities, light sensitivity, and red urine. Individuals with this disease are still anemic—a condition that can be alleviated somewhat by the heme acquired from drinking blood. This combination of traits sounds like the werewolf and vampire legends of Europe, which may have their base in this rare biochemical disease.
The process in which body proteins are continuously hydrolyzed and resynthesized is called protein turnover. The life expectancy, of body proteins is a measure of how fast they are broken down and resynthesized, expressed as a half-life. However, controlling a protein’s rate of degradation is therefore as important to the cellular and organismal economy as is controlling its rate of synthesis.
Protein degradation by non-specific mechanism
Generally “non-specific” protein degradation takes place in lysosomes – specialized organelles that operate at low pH (to denature proteins) and contain proteases for proteins, lipases for lipids, and many other hydrolases (~ 50 total). Both internal proteins (enclosed in vacuoles that fuse with lysosomes) and external proteins (obtained via endocytosis) are transported to lysosomes where proteins are degraded and the resulting amino acids either recycled for new protein synthesis or degraded for energy production or storage. In normal healthy cells, lysosomal protein degradation is nonselective. However, lysosomes in tissues that waste away or atrophy during starvation (e.g., liver and muscle, but thankfully not brain and testes) have a special degradative pathway activated only during prolonged fasting – this involves selective degradation of cytosolic proteins containing the pentapeptide sequence Lys-Phe-Glu-Arg-Gln (KFERQ), thus sparing more essential proteins from increased non-selective degradation. Such KFERQ proteins are selectively lost from tissues that atrophy in response to fasting (e.g., liver and kidney) but not from tissues that do not do so (e.g., brain and testes).
Protein degradation by specific mechanism
Although most control of metabolic activity and cell function is at the level of post-translational control or action of regulatory molecules, some control is at the actual level of the proteins themselves – by synthesis and/or degradation. This controlled or programmed protein degradation involves the Ubiquitin-Proteasome system.
This process occurs in three steps:
- In an ATP-requiring reaction, ubiquitin’s terminal carboxyl group is conjugated, via a thioester bond, to ubiquitin-activating enzyme (E1). Most organisms have only one type of E1.
- The ubiquitin is then transferred to a specific Cys sulfhydryl group on one of numerous homologous proteins named ubiquitin-conjugating enzymes.The various E2s are characterized by a ~150-residue catalytic core containing the active site Cys.
- Ubiquitin-protein ligase (E3) transfers the activated ubiquitin from E2 to a Lys epcilon-amino group of a previously bound protein, thereby forming an isopeptide bond. Cells contain many species of E3s, each of which mediates the ubiquitination (alternatively, ubiquitylation) of a specific set of proteins and thereby marks them for degradation.
In order for a protein to be efficiently degraded, it must be linked to a chain of at least four tandemly linked ubiquitin molecules in which Lys 48 of each ubiquitin forms an isopeptide bond with the C-terminal carboxyl group of the following ubiquitin .These polyubiquitin chains may contain 50 or more ubiquitin units. Ubiquitinated proteins are dynamic entities, with ubiquitin molecules being rapidly attached and removed.
Amino Acid Metabolic Pathway Metabolism is usually divided into two categories:
Catabolism, that breaks down organic matter and harvests energy by way of cellular respiration and anabolism that uses energy to construct components of cells such as proteins and nucleic acids.
Overview: Ammonia (NH3) is toxic. Waste NH3 groups (mostly from amino acids, but also from other sources) are safely incorporated into urea by the liver for disposal by the kidneys in the urine. The deaminated C-skeletons of amino acids can be oxidized for energy, stored as triglyceride until needed, or used for synthesis of new glucose (gluconeogenesis, mostly in liver). Both waste NH3 groups and C-skeletons for gluconeogenesis travel from tissues to liver as glutamine, glutamate, or alanine. Alanine carries both C-skeletons and waste NH3, both derived from skeletal muscle proteolysis, to liver where it serves as a major substrate for gluconeogenesis during fasting.
Glutamate is a central molecule in amino acid and Nitrogen (N) metabolism. Transaminases funnel waste NH3 to glutamate (a-ketoglutarate accepts the NH3 group from all other amino acids to form glutamate). Glutamate can be oxidatively deaminated to release free ammonia, which enters the urea cycle (liver). Aspartate, formed by transamination of oxaloacetate, provides the other NH3 group for urea; the C-skeleton of what used to be aspartate is eventually recycled to oxaloacetate, then transaminated to aspartate. The newly formed urea travels in the blood to the kidneys, where it is excreted in the urine. Some NH3 groups also come from deamination of glutamine in liver (for urea synthesis) and in kidney (for excretion into urine; involved in acid-base balance).
The Fate of Nitrogen (N) Atoms during catabolism of amino acids
The N atoms in amino acids are either excreted or used to synthesize other N-containing compounds.
There are three stages in nitrogen catabolism:
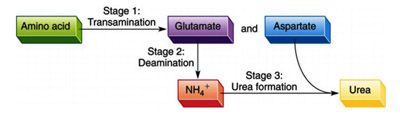
Stage 1: Transamination
Stage 2: Deamination and
Stage 3: Urea Formation.
Transamination
Most amino acids are deaminated by transamination, the transfer of their amino group to an a-keto acid to yield the a-keto acid of the original amino acid and a new amino acid. The predominant amino group acceptor is a-ketoglutarate, producing glutamate and the new a-keto acid:
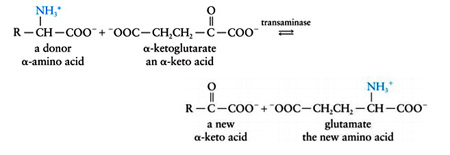
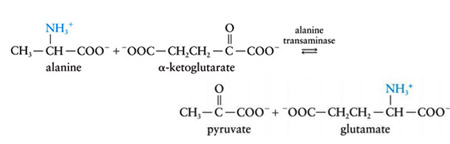
Glutamate amino group, in turn, can be transferred from alanine in a second transamination reaction, yielding pyruvate and reforming a-ketoglutarate.
The enzymes that catalyze transamination, called aminotransferases or transaminases, require the coenzyme pyridoxal-5_-phosphate (PLP). PLP is a derivative of pyridoxine (vitamin B6). The coenzyme is covalently attached to the enzyme via a Schiff base (imine) linkage formed by the condensation of its aldehyde group with the €-amino group of an enzyme Lys residue.
Another important example of transamination is the production of aspartate (which is used in urea formation) from the transfer of an amino group valine to oxaloacetate:
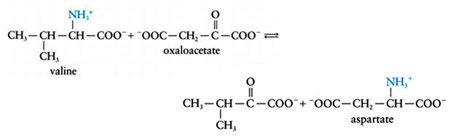
This process is an important method for the biosynthesis of the nonessential amino acids glutamate and aspartate from a variety of other amino acids.
Deamination
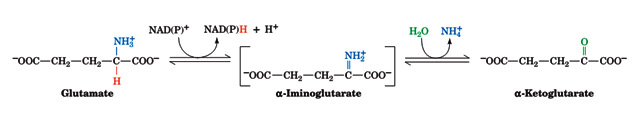
Transamination, of course, does not result in any net deamination. Glutamate, however, can be oxidatively deaminated by glutamate dehydrogenase (GDH), yielding ammonia and regenerating a-ketoglutarate for use in additional transamination reactions. Glutamate dehydrogenase, a mitochondrial enzyme, is the only known enzyme that can accept either NAD+ or NADP+ as its redox coenzyme. Oxidation is thought to occur with transfer of a hydride ion from glutamate’s Ca to NAD(P)+, thereby forming a-iminoglutarate, which is hydrolyzed to a-ketoglutarate and ammonium ion:
The enzyme is allosterically inhibited by GTP and NADH and activated by ADP and NAD+ (signaling the need to generate ATP). Because the product of the reaction, a-ketoglutarate, is an intermediate of the citric acid cycle, activation of glutamate dehydrogenase can stimulate flux through the citric acid cycle, leading to increased ATP production by oxidative phosphorylation.
The glutamate dehydrogenase reaction functions to eliminate amino groups from amino acids that undergo transamination reactions with a-ketoglutarate
Urea cycle
The urea cycle ( Ornithine cycle) is a biochemical reactions occurring in many animals that produces urea ((NH2)2CO) from ammonia(NH3). This cycle was first discovered by Hans Krebs and Kurt Henseleit in 1932. In mammals, the urea cycle takes place primarily in the liver, and to a lesser extent in the kidney.
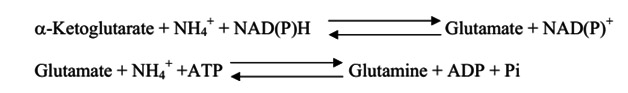
Every amino acid contains at least one amino group. Therefore every amino acid degradation pathway has a key step where the amino group is removed. In short amino acid catabolism generates ammonia. Brain tissue is very sensitive to ammonia. Ammonia intoxication produces a exhausted state. Cells get rid of excess ammonia by the reductive amination of a-ketoglutarate to form glutamate by glutamate dehydrogenase and the conversion of glutamate into glutamine by glutamine synthetase. Both of these enzymes are found in high concentrations in the brain. A high concentration of ammonia shifts the equilibrium of these two reactions towards glutamine. Glutamine synthetase use ATP to activate glutamate for amination to form glutamine. The result is high concentrations of ammonia deplete cells of ATP. it is essential for brain function.
Glutamate is a neurotransmitter and the precursor for ?-aminobutyrate (GABA) which is another important neurotransmitter. High concentrations of ammonia deplete the concentration of glutamate which produces a similar decrease of GABA which impairs brain function.
Excess amino groups are channeled in to a single excretory end product. Bony fish excrete ammonia and are thus called ammonoteilic animals. The liver of these bony fish releases ammonia into the blood stream where it is carried to the gills where it is rapidly cleared. Birds and reptiles excrete uric acid and thus are called uricotelic animals. Excretion of urea requires lots of water. If bird secreted urea, they would be weighed down by the large volume of water required and would be unable to fly. Instead they secrete uric acid in their feces. Terrestrial animals such as ourselves excrete urea and are called ureotilic animals. In ureotilic organisms, excess ammonia is channeled to the mitochondria of liver cells and converted into carbamoyl phosphate. Carbamoyl phosphate enters the nitrogen in to the urea cycle.
The Urea cycle consists of 4 reactions.
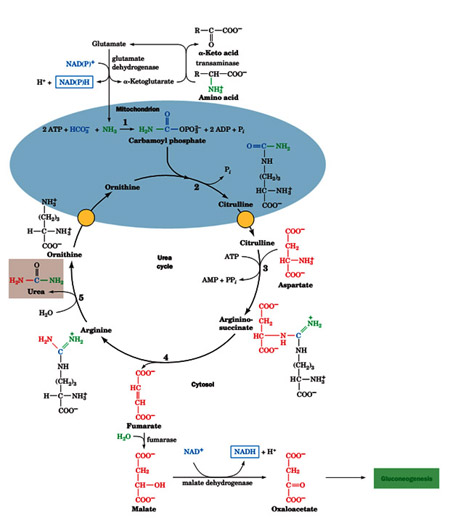
The first reaction occurs in the matrix of the mitochondria. The subsequent reactions occur in the cytosol. This is a pathway that spans two cellular compartments. The first reaction is catalyzed by ornithine trans carbamoylase which transfers a carbamoyl group from carbamoyl phosphate to ornithine to form citrulline.
The second reaction is catalyzed by argininosuccinate synthetase. This enzyme uses ATP to activate citrulline by forming a citrullyl-AMP intermediate. This intermediate is attacked by the amino group of an aspartate residue to formargininosuccinate. The third step is catalyzed by argininosuccinate lyase which cleaves argininosuccinate into fumarate and arginine. The last step is catalyzed by arginase which cleaves arginine to produce urea and ornithine completing the cycle.
The first step of the urea cycle begins in the mitochondrial matrix. Ammonia is produced in the mitochondrial matrix by the enzymatic activities of glutaminase and glutamate dehydrogenase. Carbamoyl phosphate synthetase I is the enzyme that takes the ammonia, bicarbonate and 2 molecules of ATP to produce carbamoyl phosphate. This enzyme activates bicarbonate by the same method used by biotin containing enzymes. In fact carbamoyl phosphatesynthetase is homologous to the biotin family of enzymes. Carbamoyl phosphate synthetase I activates bicarbonate by phosphorylation with ATP to form carboxyphosphate. Ammonia then reacts with carboxyphosphate to from a carbamate intermediate. A second molecule of ATP is used to phosphorylate the carbamate intermediate to form carbamoyl phosphate. Carbamoyl phosphate synthetase I is the first committed step of the urea cycle.
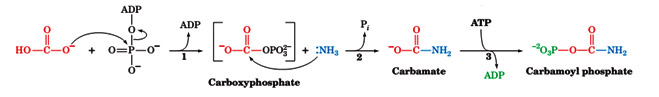
The carbamoyl phosphate formed by this reaction enters into the urea cycle as a substrate for ornithine trans carbamoylase which catalyzes the following reaction.
Amino Acid Metabolic Pathway Metabolism is usually divided into two categories:
Catabolism, that breaks down organic matter and harvests energy by way of cellular respiration and anabolism that uses energy to construct components of cells such as proteins and nucleic acids.
Overview: Ammonia (NH3) is toxic. Waste NH3 groups (mostly from amino acids, but also from other sources) are safely incorporated into urea by the liver for disposal by the kidneys in the urine. The deaminated C-skeletons of amino acids can be oxidized for energy, stored as triglyceride until needed, or used for synthesis of new glucose (gluconeogenesis, mostly in liver). Both waste NH3 groups and C-skeletons for gluconeogenesis travel from tissues to liver as glutamine, glutamate, or alanine. Alanine carries both C-skeletons and waste NH3, both derived from skeletal muscle proteolysis, to liver where it serves as a major substrate for gluconeogenesis during fasting.
Glutamate is a central molecule in amino acid and Nitrogen (N) metabolism. Transaminases funnel waste NH3 to glutamate (a-ketoglutarate accepts the NH3 group from all other amino acids to form glutamate). Glutamate can be oxidatively deaminated to release free ammonia, which enters the urea cycle (liver). Aspartate, formed by transamination of oxaloacetate, provides the other NH3 group for urea; the C-skeleton of what used to be aspartate is eventually recycled to oxaloacetate, then transaminated to aspartate. The newly formed urea travels in the blood to the kidneys, where it is excreted in the urine. Some NH3 groups also come from deamination of glutamine in liver (for urea synthesis) and in kidney (for excretion into urine; involved in acid-base balance).
The Fate of Nitrogen (N) Atoms during catabolism of amino acids
The N atoms in amino acids are either excreted or used to synthesize other N-containing compounds.
There are three stages in nitrogen catabolism:
Stage 1: Transamination
Stage 2: Deamination and
Stage 3: Urea Formation.
Transamination
Most amino acids are deaminated by transamination, the transfer of their amino group to an a-keto acid to yield the a-keto acid of the original amino acid and a new amino acid. The predominant amino group acceptor is a-ketoglutarate, producing glutamate and the new a-keto acid:
Glutamate amino group, in turn, can be transferred from alanine in a second transamination reaction, yielding pyruvate and reforming a-ketoglutarate.
The enzymes that catalyze transamination, called aminotransferases or transaminases, require the coenzyme pyridoxal-5_-phosphate (PLP). PLP is a derivative of pyridoxine (vitamin B6). The coenzyme is covalently attached to the enzyme via a Schiff base (imine) linkage formed by the condensation of its aldehyde group with the €-amino group of an enzyme Lys residue.
Another important example of transamination is the production of aspartate (which is used in urea formation) from the transfer of an amino group valine to oxaloacetate:
This process is an important method for the biosynthesis of the nonessential amino acids glutamate and aspartate from a variety of other amino acids.
Deamination
Transamination, of course, does not result in any net deamination. Glutamate, however, can be oxidatively deaminated by glutamate dehydrogenase (GDH), yielding ammonia and regenerating a-ketoglutarate for use in additional transamination reactions. Glutamate dehydrogenase, a mitochondrial enzyme, is the only known enzyme that can accept either NAD+ or NADP+ as its redox coenzyme. Oxidation is thought to occur with transfer of a hydride ion from glutamate’s Ca to NAD(P)+, thereby forming a-iminoglutarate, which is hydrolyzed to a-ketoglutarate and ammonium ion:
The enzyme is allosterically inhibited by GTP and NADH and activated by ADP and NAD+ (signaling the need to generate ATP). Because the product of the reaction, a-ketoglutarate, is an intermediate of the citric acid cycle, activation of glutamate dehydrogenase can stimulate flux through the citric acid cycle, leading to increased ATP production by oxidative phosphorylation.
The glutamate dehydrogenase reaction functions to eliminate amino groups from amino acids that undergo transamination reactions with a-ketoglutarate
Urea cycle
The urea cycle ( Ornithine cycle) is a biochemical reactions occurring in many animals that produces urea ((NH2)2CO) from ammonia(NH3). This cycle was first discovered by Hans Krebs and Kurt Henseleit in 1932. In mammals, the urea cycle takes place primarily in the liver, and to a lesser extent in the kidney.
Every amino acid contains at least one amino group. Therefore every amino acid degradation pathway has a key step where the amino group is removed. In short amino acid catabolism generates ammonia. Brain tissue is very sensitive to ammonia. Ammonia intoxication produces a exhausted state. Cells get rid of excess ammonia by the reductive amination of a-ketoglutarate to form glutamate by glutamate dehydrogenase and the conversion of glutamate into glutamine by glutamine synthetase. Both of these enzymes are found in high concentrations in the brain. A high concentration of ammonia shifts the equilibrium of these two reactions towards glutamine. Glutamine synthetase use ATP to activate glutamate for amination to form glutamine. The result is high concentrations of ammonia deplete cells of ATP. it is essential for brain function.
Glutamate is a neurotransmitter and the precursor for ?-aminobutyrate (GABA) which is another important neurotransmitter. High concentrations of ammonia deplete the concentration of glutamate which produces a similar decrease of GABA which impairs brain function.
Excess amino groups are channeled in to a single excretory end product. Bony fish excrete ammonia and are thus called ammonoteilic animals. The liver of these bony fish releases ammonia into the blood stream where it is carried to the gills where it is rapidly cleared. Birds and reptiles excrete uric acid and thus are called uricotelic animals. Excretion of urea requires lots of water. If bird secreted urea, they would be weighed down by the large volume of water required and would be unable to fly. Instead they secrete uric acid in their feces. Terrestrial animals such as ourselves excrete urea and are called ureotilic animals. In ureotilic organisms, excess ammonia is channeled to the mitochondria of liver cells and converted into carbamoyl phosphate. Carbamoyl phosphate enters the nitrogen in to the urea cycle.
The Urea cycle consists of 4 reactions.
The first reaction occurs in the matrix of the mitochondria. The subsequent reactions occur in the cytosol. This is a pathway that spans two cellular compartments. The first reaction is catalyzed by ornithine trans carbamoylase which transfers a carbamoyl group from carbamoyl phosphate to ornithine to form citrulline.
The second reaction is catalyzed by argininosuccinate synthetase. This enzyme uses ATP to activate citrulline by forming a citrullyl-AMP intermediate. This intermediate is attacked by the amino group of an aspartate residue to formargininosuccinate. The third step is catalyzed by argininosuccinate lyase which cleaves argininosuccinate into fumarate and arginine. The last step is catalyzed by arginase which cleaves arginine to produce urea and ornithine completing the cycle.
The first step of the urea cycle begins in the mitochondrial matrix. Ammonia is produced in the mitochondrial matrix by the enzymatic activities of glutaminase and glutamate dehydrogenase. Carbamoyl phosphate synthetase I is the enzyme that takes the ammonia, bicarbonate and 2 molecules of ATP to produce carbamoyl phosphate. This enzyme activates bicarbonate by the same method used by biotin containing enzymes. In fact carbamoyl phosphatesynthetase is homologous to the biotin family of enzymes. Carbamoyl phosphate synthetase I activates bicarbonate by phosphorylation with ATP to form carboxyphosphate. Ammonia then reacts with carboxyphosphate to from a carbamate intermediate. A second molecule of ATP is used to phosphorylate the carbamate intermediate to form carbamoyl phosphate. Carbamoyl phosphate synthetase I is the first committed step of the urea cycle.
The carbamoyl phosphate formed by this reaction enters into the urea cycle as a substrate for ornithine trans carbamoylase which catalyzes the following reaction.
- There is a citrulline transporter that transports citrulline from the matrix of the mitochondria to the cytosol. There is also an ornithine trasporter that transport ornithine from the cytosol into the matrix of the mitochondria. The rest of the reactions of the urea cycle occur in the cytosol.
- The next step of the urea cycle is catalyzed by argininosuccinate synthetase. This enzyme uses ATP to activate citrulline by forming a citrully-AMP intermediate
- In the second half of the argininosuccinate synthetase reaction, the a-amino group of aspartate attacks the imino carbon releasing AMP and producing argininosuccinate.
- The third step of the urea cycle is catalyzed by arginino succinate lyase. The products of this reaction are arginine and fumarate. The fumarate product is an important link between the urea cycle and the citric acid cycle.
- The last reaction of the urea cycle is catalyzed by arginase which catalyzes the hydrolysis of guanidino group to produce urea and ornithine. Ornthine is then transported back in the matrix of the mitochondria and the cycle is complete. There are two nitrogens in urea. One came from the ammonia produced in the matrix in the mitochondria captured in the form of carbamoyl phosphate. The second nitrogen was the a-amino group of the aspartate substrate in the argininosuccinate synthetase reaction. The sole carbon of urea was originally the carbon of the bicarbonate.
In the mitochondria oxaloacetate is tranaminated by glutamate by aspartate amino transferase to produce the aspartate which is then transported into the cytosol where it is required for the argininosuccinate synthetase reaction. These reactions together are called the aspartate-argininosuccinate shunt. The summary of the links between the urea cycle and the citric acid cycle is shown above diagram.
Regulation of urea cycle
- Carbamoyl phosphate synthetase I, which catalyzes the first committed step of the urea cycle, is allosterically activated by N-acetylglutamate. This metabolite is synthesized from glutamate and acetyl-CoA by Nacetyl glutamate synthase. When increase the rates of breakdown of amino acid, the concentration of glutamate increases as a result of transamination. The increased glutamate stimulates N-acetylglutamate synthesis. The resulting activation of carbamoyl phosphate synthetase increases the rate of urea production. Thus, the excess nitrogen produced by amino acid breakdown is efficiently excreted.
- The remaining enzymes of the urea cycle are controlled by the concentrations of their substrates. In individuals with inherited deficiencies in urea cycle enzymes other than arginase, the corresponding substrate builds up, increasing the rate of the deficient reaction so that the rate of urea production is normal.
Free amino acids are not stored. Therefore, in all organisms, amino acid breakdown occurs whenever amino acid levels exceed requirements for synthetic processes Amino acid catabolism is critically important. There are two reasons for this: 1) amino acids are a potential source of energy, and especially during fasting, of glucose, and 2) incomplete metabolism of a number of amino acids results in accumulation of toxic amino acid breakdown intermediates. When considering their degradation, amino acids are generally divided into two classes. The term glucogenic refers to amino acids with a carbon skeleton that can be converted to a gluconeogenic or TCA cycle intermediate. These amino acids can be used to synthesize glucose. The term ketogenic refers to amino acids with a carbon skeleton that can only be converted to acetyl CoA, to acetoacetyl-CoA, or to acetoacetate. The ketogenic amino acids are a potential source of ketone bodies (hence the name “ketogenic”), but cannot be used to synthesize glucose. Some amino acids fall into both categories. The diagram below summarizes the conversion of the twenty standard amino acids into compounds that can be used for energy. Note that some of the amino acids are converted into more than one metabolite; these amino acids are indicated by an asterisk in the figureThe amino acids are also frequently divided into families, based on the type of amino acid, or the final product formed from the amino acid.
Synthesis of amino acids
Synthesis and/or collection of amino acids is critical for cell survival. They not only serve as the building blocks for proteins but also as starting points for the synthesis of many important cellular molecules including vitamins and nucleotides. In most cases bacteria would rather use amino acids in their environment than make them from scratch. It takes a considerable amount of energy to make the enzymes for the pathway as well as the energy required to drive some of the reactions of amino acid biosynthesis. The genes that code for amino acid synthesis enzymes and the enzymes themselves are under tight control and are only turned on when they are needed.The amino acids synthesis pathways can be grouped into several logical units. These units reflect either common mechanisms or the use of common enzymes that synthesize more than one amino acid. These categories are: simple reactions, branch chain amino acids, aromatic amino acids, threonine/lysine, serine/glycine, and unique pathways. The aromatic amino acids, threonine/lysine and serine/glycine pathways have a common beginning and then diverge to form the amino acid of interest.
Notice that each pathway begins with a central metabolite or something derived from "central metabolism". Using common compounds instead of synthesizing them from scratch saves energy and conserves genes since fewer enzymes are needed to code for the pathways. Most of standard amino acids are synthesized by glycolysis and citric acid cycle is shown below.
Inborn errors of amino acid metabolism
An inability to degrade amino acids causes many genetic diseases in humans. These diseases include phenylketonuria (PKU), which results from an inability to convert phenylalanine to tyrosine. The phenylalanine is instead transaminated to phenylpyruvic acid, which is excreted in the urine, although not fast enough to prevent harm. PKU was formerly a major cause of severe mental retardation. Now, however, public health laboratories screen the urine of every newborn child in the United States for the presence of phenylpyruvate, and place children with the genetic disease on a synthetic low-phenylalanine diet to prevent neurological damage.
Heme biosynthesis
Inborn metabolic diseases that interfere with heme biosynthesis are called porphyrias. Porphyrias have a variety of symptoms. A deficiency in the enzyme responsible for the condensation of porphobilinogen to the 4-membered ring system leads to a condition called acute intermittent porphyria, which is characterized by occasional episodes of abdominal pain and psychiatric symptoms. Defects in the later enzymes of the pathway lead to an excess accumulation of the uroporphobilinogens in the tissues, where they cause a variety of symptoms, including hairy skin, skeletal abnormalities, light sensitivity, and red urine. Individuals with this disease are still anemic—a condition that can be alleviated somewhat by the heme acquired from drinking blood. This combination of traits sounds like the werewolf and vampire legends of Europe, which may have their base in this rare biochemical disease.
Published date : 11 Jun 2014 12:43PM




More Articles


